Hepatitis Delta Virus Detected in Salivary Glands of Sjögren’s Syndrome Patients and Recapitulates a Sjögren’s Syndrome-Like Phenotype in Vivo
Published May 23, 2016
Read in PDF format
Standfirst
Hepatitis delta virus has been detected in a subset of Sjögren’s syndrome patients and shows capacity to trigger a similar disease phenotype in vivo.
Authors
Melodie L. Weller1, Matthew R. Gardener1, Zoe C. Bogus1, Michael A. Smith1, Elisa Astorri 2, Drew G.
Michael1, Donald A. Michael1, Changyu Zheng1, Peter D. Burbelo3, Zhennan Lai1, Paul A. Wilson 4, William Swaim1, Beverly Handelman1, Sandra A. Afione1, Michele Bombardieri2, John A.
Chiorini1
Affiliated Institutions
1
Molecular Physiology and Therapeutics Branch, National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, Maryland
2
Centre for Experimental Medicine & Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine & Dentistry, Queen Mary
University of London, London, UK
3
Dental Clinical Research Core, National Institute of Dental and Craniofacial Research, National Institutes of Health, Bethesda, Maryland
4
National Intramural Database, Division of Enterprise and Custom Applications, Center for Information Technology, National Institutes of Health, Bethesda,
Maryland
Corresponding Authors
Melodie L. Weller
wellerm@mail.nih.gov
240-422-7857
John A. Chiorini
jchiorini@dir.nidcr.nih.gov
DOI
10.20411/pai.v1i1.72
Suggested Citation
Weller ML, Gardener MR, Bogus ZC, Smith MA, Astorri E, Michael DG, Michael DA, Zheng C, Burbelo PD, Lai Z, Wilson PA, Swaim W, Handelman B, Afione SA, Bombardieri M, Chiorini JA. Hepatitis Delta Virus Detected in Salivary Glands of Sjögren’s Syndrome Patients and Recapitulates a Sjögren’s Syndrome-Like Phenotype in Vivo. Pathogens and Immunity. 2016;1(1):12-40. 10.20411/pai.v1i1.72
Abstract
Background:
Low-level, chronic viral infections have been suspect in the development of select autoimmune diseases, including primary Sjögren’s syndrome (pSS).
Multiple studies have shown stimulation of antiviral response pathways in pSS tissues suggestive of a viral infection. Yet, with this data in hand, a
causal link between a viral infection and development of pSS had not been identified. Therefore, a study was designed to further define the viral landscape
within pSS-affected salivary gland tissue to identify potential viral-mediated triggers in the pathogenesis of this autoimmune disease.
Methods:
A viral microarray was utilized to measure viral transcripts present in salivary gland tissue from patients diagnosed with pSS compared to healthy
controls. Murine models of salivary gland localized HDV antigen expression were developed to evaluate the capacity of a chronic HDV signature to trigger
the development of a pSS-like phenotype.
Results:
Through this analysis, two distinct viral profiles were identified, including the increased presence of hepatitis delta virus (HDV) in 50% of pSS patients
evaluated. Presence of HDV antigen and sequence were confirmed in minor salivary gland tissue. Patients with elevated HDV levels in salivary gland tissue
were negative for detectible hepatitis B virus (HBV) surface antigen and antibodies to HBV or HDV. Expression of HDV antigens in vivo resulted in reduced
stimulated saliva flow, increase in focal lymphocytic infiltrates, and development of autoantibodies.
Conclusion:
Identification of HDV in pSS patients and induction of a complete pSS-like phenotype in vivo provides further support of a viral-mediated etiopathology in
the development of pSS.
Keywords: Sjogren’s syndrome, Sjögren’s syndrome, Viral-mediated autoimmunity, Hepatitis Delta Virus, Hepatitis D Virus, HDV, xerostomia, xerophthalmia, Sjogrens syndrome, Sjögrens syndrome
Introduction
Viral infections are thought to play a role in the development of chronic diseases, including autoimmune diseases [1]. While evidence of an antiviral response has been observed in multiple autoimmune diseases [2-8], association with a viral agent has not always been clear. This may be attributed to a multifactorial etiology of chronic autoimmunity requiring a
genetic susceptibility component in combination with a persistent viral infection or chronic viral exposure to trigger disease pathogenesis. Therefore, a
study was designed to further define the viral landscape and potential viral triggers of autoimmunity in disease-affected tissue as illustrated in patients
with primary Sjögren’s syndrome.
Primary Sjögren’s syndrome (pSS) is an autoimmune disease estimated to affect over 35 million people worldwide [9,10]. This autoimmune disease is currently diagnosed by a reduction in tear and/or saliva secretion, accumulation of focal lymphocytic infiltrates in salivary
gland tissue, and development of antibodies against Ro(SSA), La(SSB), and nuclear proteins [11]. Epidemiological studies have reported patients experiencing subjective xerostomia and development of autoantibodies over a decade prior to diagnosis,
suggesting a chronic, slowly progressing development of the disease pathology [12-14]. While the underlying trigger(s) of Sjögren’s syndrome remains to be defined, various studies have pointed to an altered genetic susceptibility in
connection with environmental exposures, including viral infections.
A common antiviral signature in combination with divergent symptomology suggests a multivariate etiology underlying the Sjögren’s syndrome phenotype.
Clinical studies have reported unique groups within the pSS patient population presenting with different symptom profiles and disease progression based on
age of onset, sex, immunological presentation, and extraglandular involvement across a large cohort of pSS patients [15]. Gene expression analyses within affected salivary gland tissue have identified stimulation of antiviral response pathways, including upregulation of
type I interferon (IFN) inducible genes and upstream viral-sensing Toll-like receptors [8,16-18]. Although these studies echo a similar overlying antiviral response, the divergent clinical characteristics across the pSS patient population may be due
to multiple etiologies, and potentially different viral infections, behind the collective phenotypic presentation of primary Sjögren’s syndrome. Prior
studies have detected the presence of viruses in salivary glands of Sjögren’s syndrome patients, including Epstein-Barr Virus (EBV), Coxsackie virus, and
Human T-lymphotropic virus (HTLV-1) [19-22]. Yet, even with the discovery of these viruses in Sjögren’s syndrome patients, a clear connection between these viral infections and the development of
primary Sjögren’s syndrome has been difficult to establish.
To globally identify potential viral triggers in the pathogenesis of Sjögren’s syndrome, a custom viral microarray was designed to identify low-level
transcripts from actively replicating viruses present within salivary gland tissue of primary Sjögren’s syndrome patients compared to healthy controls.
Through this analysis, multiple virus profiles were identified in the pSS patient cohort evaluated. One of the virus profiles identified included the
presence of hepatitis delta virus (HDV). Taking the next step, we evaluated the capacity of HDV antigens to trigger a Sjögren’s syndrome-like phenotype in
vivo.
Materials and Methods
Patients and samples
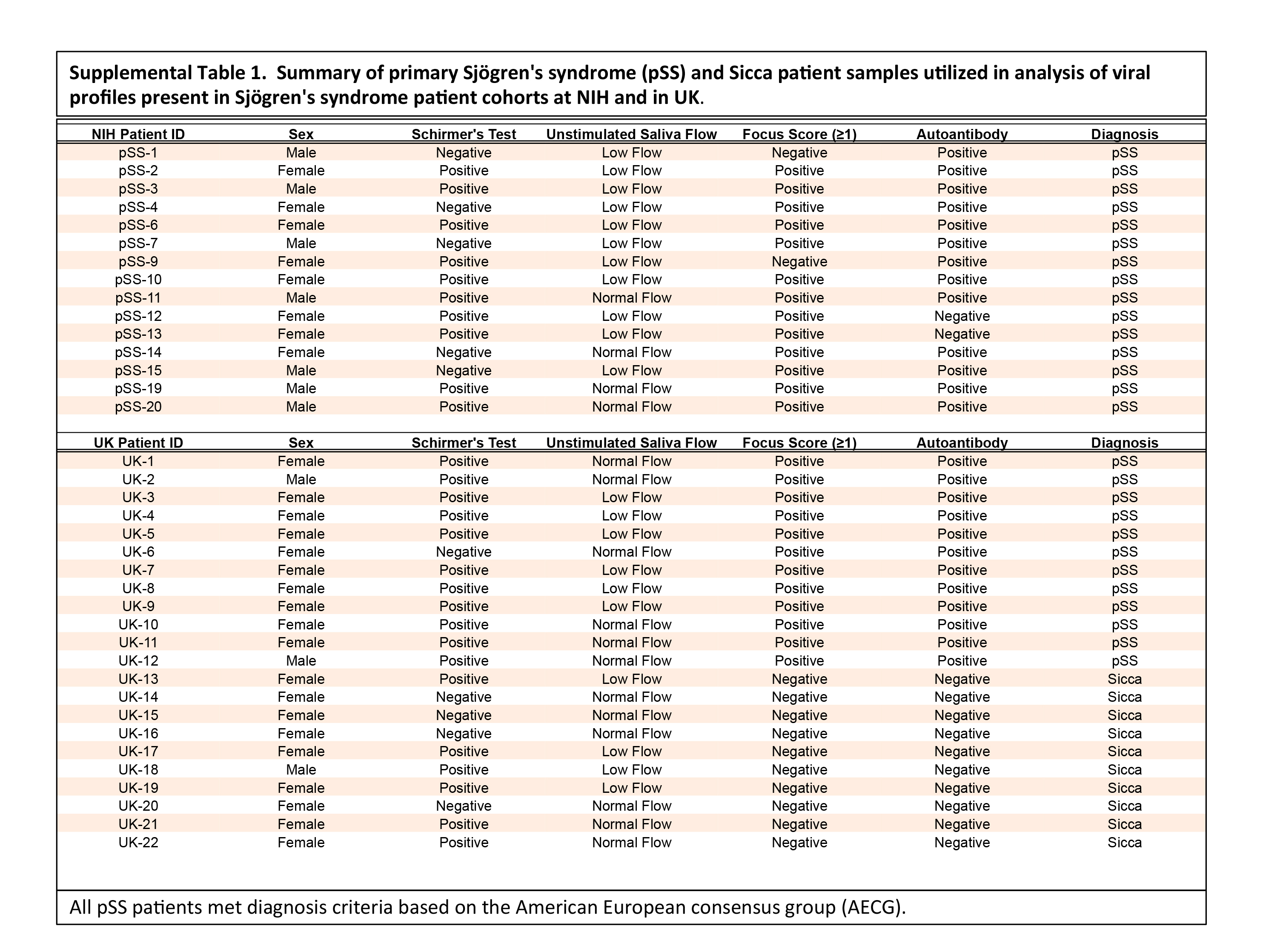
All patient studies were reviewed and approved by the Institutional Review Board USA (NCT00001390) and all patients signed a written consent form prior to
enrollment. All primary Sjögren’s syndrome patients met American-European Consensus Group (AECG) criteria [11] for diagnosis of Sjögren’s syndrome. Clinical parameters and serum and salivary gland labial biopsies from 14 healthy controls and 15 patients diagnosed
with primary Sjögren’s syndrome were utilized in this study and are summarized in Supplemental Table 1.
Patients from the second United Kingdom (UK) cohort was approved by the hospital Ethics Committee (REC 05/Q0702/1) and all patients signed a written
informed consent. This second dataset, summarized in Supplemental Table 1, included RNA samples isolated from labial salivary gland biopsies obtained from
a total of 12 pSS patients and minor salivary gland biopsies from 10 sicca patients. Sicca samples were defined as patients experiencing select
characteristics of pSS but did not meet full criteria for diagnosis of Sjögren’s syndrome at time of tissue acquisition. An additional confirmation study,
consisting of 8 pSS and 8 sicca samples, including samples (two positive and two negative) overlapping with those tested in the second phase of the study
conducted at the NIH, were analyzed for independent confirmation of HDV in RNA isolated from minor salivary gland biopsies conducted at the UK location.
Microarray
Viral microarray probes were designed using a modified method reported by Wang et al. [23]. Briefly, viral genomes were fragmented into 70nt sequences and iterated across 15nt offsets to produce up to 30,000 candidate probes per viral sequence.
The resulting 70mer fragments were then blasted against the RefSeq nucleotide database, and probes with E-values < 0.05 were retained. These
70mer sequences were further filtered using Agilent eArray parameters for efficient melting temperature and nucleotide composition rendering optimized
60mer viral microarray probe sequences. A maximum of 5 probes per viral sequence was retained. Each array contained over 3000 probes that recognized
putative conserved regions in viruses known to infect animals and covered 31 viral families. RNA isolated from minor salivary gland tissue was processed as
previously reported [24].
Genespring (v.12.0) software was utilized for viral microarray analysis. Microarray data were normalized using quantile normalization and filtered for
background intensity (threshold set to 25% of any one group having a raw probe intensity of > 100). Dataset evaluated in this study have been deposited
in NCBI GEO Database (series no. GSE77599). Collective analysis identified probes significantly increased across the collective pSS patient group compared
to the collective healthy controls and that presented a fold change > 2.0 in pSS relative to healthy controls. Statistical significance was corrected for
multiple comparisons using the Benjamini-Hochberg correction method. Subgroup analysis was performed through identifying normalized the probe intensity of
each probe and identifying probes for each individual patient that had a greater than a 95% confidence interval of the probe intensity for the collective
healthy controls. Probes selected for further analysis presented with intensities greater than the 95% confidence interval of healthy controls and were
present in at least 25% of the pSS patient samples tested. Pairwise correlations between normalized probe intensities of probes present in subgroups of pSS
were then identified and mapped using JMP (v.10.0) statistical software from SAS. Heatmap and cluster analysis were generated using CIMminer (NCI).
PCR detection of HDV sequence
Reverse transcription of RNA isolated from minor salivary gland biopsies was performed using Life Technologies Superscript II and random hexamer primers as
per the manufacturer’s specifications. The genomic HDV sequence was identified in RNA using a modified nested PCR classically used in the detection of HDV
in HBV:HDV co-infections [25]. Modification to protocol included the addition of dNTPs to the first round of nested PCR. The transcript targeted nested RT-qPCR design used to detect
both HDV genomic and transcript sequences and, in our testing, was more sensitive than the Smedile et al. PCR used to detect only the HDV genome.
The first round PCR was performed using Qiagen HotStarTaq DNA Polymerase as follows: 5μl of Q-solution; 3.5μl of 25 mM MgCl2; 2.5μl of 10X PCR buffer;
0.25μl Hot Start enzyme; 0.25μl 10mM dNTPs; 0.125μl 100mM tHDV-1F (5′- GGCTACTCTTCTTTCCCTTCTC-3′) and 0.125μl 100mM tHDV-1R (5′- ACAAGGAGAGGCAGGATCA-3′);
1μl cDNA; and PCR grade water to 25μl total volume. First round PCR conditions: 1 cycle of 5:00 minutes at 95°C; 35 cycles of 0:30 minutes at 94°C; 0:30
minutes at 50°C; 0:30 minutes at 72°C; and 1 cycle of 10:00 minutes at 72°C. The second round of qPCR was performed using the following primer and probe set:
tHDV-2F (5′- TCTCGTCTTCCTCGGTCAA-3′); tHDV-2R (5′-GCCCTCGAGAACAAGAAGAA -3′); and tHDV-probe (5′-FAM/TTCCTCCTTGCTGAGGTTCTTGCC/3′-TAMSp). The qPCR reaction
was performed on an Applied Biosystems ABI 7700 instrument and the reaction contained Applied Biosystems (ABI) TaqMan 2X PCR master mix, 500nM of primers,
250nM of probe, 1-3μl of first round PCR reaction, and PCR grade water to a total volume of 25μl. Increasing the volume of first-round PCR added to the
nested reaction may be required to quantify lower levels of HDV sequence present in samples. The qPCR reaction was performed as follows: 1 cycle at 50°C
for 2:00 minutes; 1 cycle at 95°C for 10:00 minutes; 40 cycles of 0:15 minutes at 95°C, 0:30 minutes at 50°C, and 1:00 minutes at 60°C. Quantitation of
GAPDH was performed using Applied Biosystems (ABI) FAM/MGB labeled probe to human GAPDH (catalog # HS99999905_m1) and an assay was performed as outlined in
the manufacturer’s suggested protocol. HDV levels were calculated as fold change in pSS relative to average of healthy controls as per ABI protocol [26]. A more detailed protocol is available upon request.
Detection of HDAg expression in salivary gland tissue
Immunohistochemical analysis of HDV antigens (HDAg) was performed using rabbit anti-HDAg antiserum as previously characterized [27,28] for human salivary gland tissues. Staining of formalin fixed paraffin embedded (FFPE) human salivary gland tissue was performed using citrate (10mM
sodium citrate, pH6.0, 0.05% Tween20) antigen retrieval, followed by 30 minute incubation in blocking solution (3% BSA in TRIS buffered saline [TBS]) at
room temperature, incubation with primary antibody overnight at 4°C (1:400 for rabbit antiserum) and 1 hour room temperature incubation in labeled
secondary antibody solution (Alexa Fluor 488 Goat Anti-Rabbit IgG, 1:500, Life Technologies). Slides were then mounted in Fluoromount G (Electron
Microscopy Systems) containing DAPI counterstain. B-cell, (B220, Abcam catalog #AB64100, 1:250) and T-cells (CD3, Abcam, catalog #AB16669, 1:100) were
detected in FFPE mouse tissues using the manufacturer’s recommended conditions. Secondary species-specific antibodies for B-cell b220 (Alexa Fluor 594 Goat
Anti-Rat IgG, 1:500, Life Technologies) and T-cell CD3 (Alexa Fluor 488 Goat Anti-Rabbit IgG, 1:500, Life Technologies) were incubated for 1 hour at room
temperature and mounted in Fluoromount G (Electron Microscopy Systems) containing DAPI counterstain.
Detection of antibody profiles by ELISA
ELISAs from International Immunodiagnostics (Foster City, CA) to detect antibodies to HDAg (catalog #277) and Abnova Corporation to detect antibodies to
HBV core protein (catalog #KA0288) were performed on serum from healthy controls and pSS patients as per manufacturer’s suggested protocols. A luciferase
immunoprecipitation system (LIPS) was performed to detect HBV core protein or HDV antigen tagged with Renilla luciferase and was performed as
previously published [29,30]. A human anti-HDAg antibody was used as the positive control (gift from John Casey, PhD, Georgetown University).
Anti-SSA/Ro, anti-SSB/La, and antinuclear antibodies (ANA) were detected by ELISA from Alpha Diagnostics International using human serum as per the
manufacturer’s protocol. Total IgG (eBiosences), anti-SSA/Ro (Alpha Diagnostics International), anti-SSB/La (Alpha Diagnostics International), and ANA
(Alpha Diagnostics International) were detected in mouse serum by ELISA as per the manufacturer’s suggested protocols.
Animal Model
All animal studies were approved by the NIDCR Institutional Animal Care and Use Committee (IACUC) and performed in compliance with the NIH Guide for the
Care and Use of Laboratory Animals. Recombinant adeno-associated virus serotype 2 (AAV2) was produced and utilized for cannulation of submandibular
salivary glands in 8-week-old female C57BL/6 mice as previously reported [31]. Mice were cannulated with 1.0 x 1010 genomic particle/gland AAV2 containing S-HDAg or L-HDAg sequences and spiked with AAV containing
luciferase transgene as a control for cannulation efficacy. The combined expression of S-HDAg and L-HDAg (S-HDAg/L-HDAg) was facilitated by delivery of a
1:1 mixture of AAV containing S-HDAg or L-HDAg. Control mice were cannulated with AAV containing luciferase transgene. Viral aliquots of rAAV2-HDAg used
for cannulation were spiked with 10% rAAV2-luciferase to confirm effective cannulation. One week post-cannulation, mice were monitored for luciferase
expression in the salivary gland tissue region as previously reported [32]. Mice that had detectible levels of luciferase activity were utilized for the study and were assessed for pilocarpine stimulated saliva flow, antibody
development, lymphocytic foci development and HDAg expression at 4 months post-cannulation using the methodology previously reported [24].
Results
Viral microarray analysis was performed using RNA isolated from minor salivary gland tissue from 15 primary Sjögren’s syndrome patients and 14 healthy
controls (Supplemental Table 1). The viral microarray contained over 3000 probes for viral families known to infect animals. Probes were designed to detect
homologous sequences shared between multiple viral family members, enabling the detection of viral signatures with a limited number of probes [23,33]. This method has the potential to identify transcripts of actively replicating RNA and DNA viruses within the affected salivary gland tissue. Our
hypothesis was that a viral-mediated pSS-like phenotype may be caused by more than one type of viral infection. Therefore, the analysis of the viral array
data was performed using two different approaches: (1) identification of the collective pSS patient cohort viral signature compared to the healthy
controls; and (2) identification of individual viral signatures to identify subgroups within the pSS patient cohort compared to healthy controls.
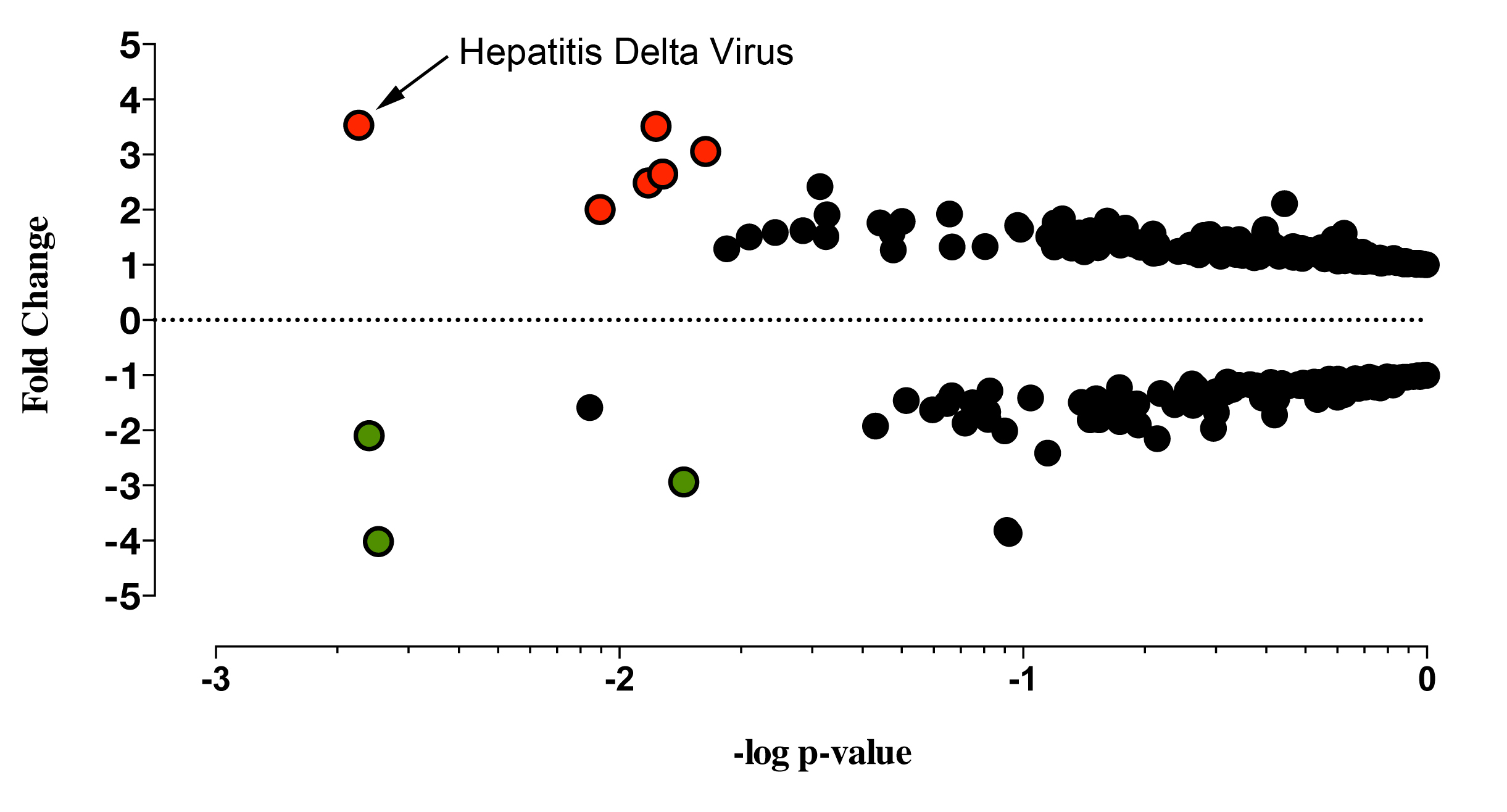
Figure 1. Collective analysis identified hepatitis delta virus in primary Sjögren’s syndrome salivary gland tissue.
Collective analysis comparing viral profiles of the pSS patient population to the healthy controls’ viral profile identified 9 probes differentially
expressed between the pSS population and the healthy controls. The graph denotes fold change and P-values (t-test) for probes above the
background threshold. Red (positive fold change) and green (negative fold change) circles denote probes rendering a P-value of < 0.05 after
Benjamini-Hochberg correction (P(Corr) < 0.05, t-test, Benjamini-Hochberg correction, n = 29). Black circles denote the remaining probes without
a significant change between pSS patients and healthy controls after Benjamini-Hochberg correction (Supplemental Table 2).
Download as a PowerPoint Slide
Identified Viral Profiles in pSS
The collective analysis of the viral transcripts differentially expressed between the pSS patient cohort and healthy controls identified 9 probes from 8
distinct viral families that were significantly altered in the pSS cohort. Six probes recognizing HDV, Herpesviridae, Retroviridae, Astroviridae,
Adenoviridae, and Circoviridae viral families were significantly increased in pSS salivary gland tissue compared to healthy controls (Figure 1,
Supplemental Table 2). Three probes recognizing Flaviviridae and Poxviridae viral families were significantly decreased in pSS compared to healthy
controls. Through this collective analysis, HDV was identified as the most significant virus profile, presenting with the highest probe intensity and
significant increase across the collective patient population compared to healthy controls.
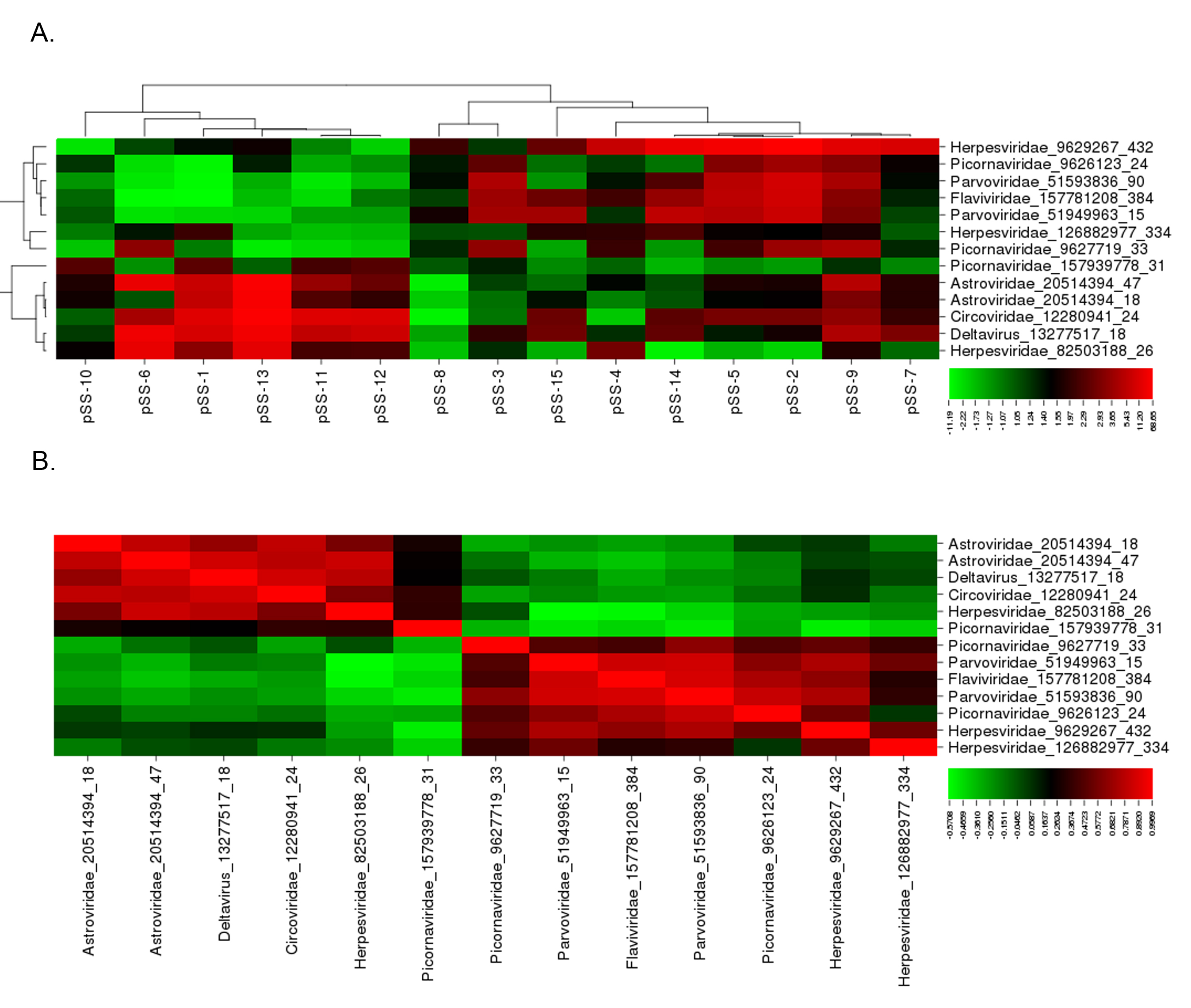
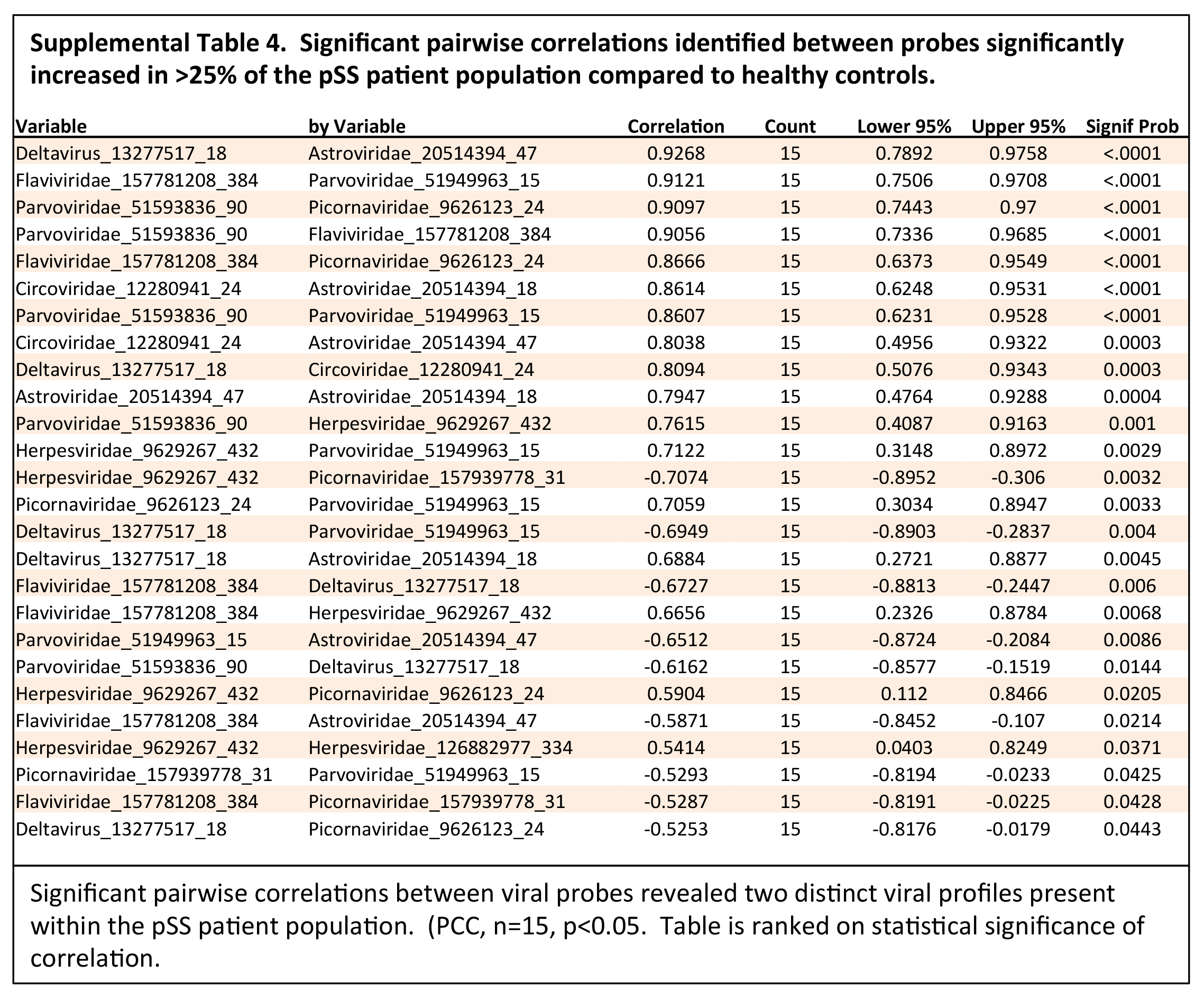
Figure 2. Subgroup analysis identified 2 distinct viral profiles present in the pSS patient population tested.
(A) Cluster analysis of the 13 probes identified through subgroup analysis rendered 2 virus profiles. Virus profile I contained probes for deltavirus,
Astroviridae, Circoviridae, and Herpesviridae. Virus profile II contained probes for Flaviviridae, Picornaviridae, Parvoviridae, and Herpesviridae. (B)
Pairwise correlation of probes identified through subgroup analysis further defined the 2 divergent virus profiles.
Download as a PowerPoint Slide
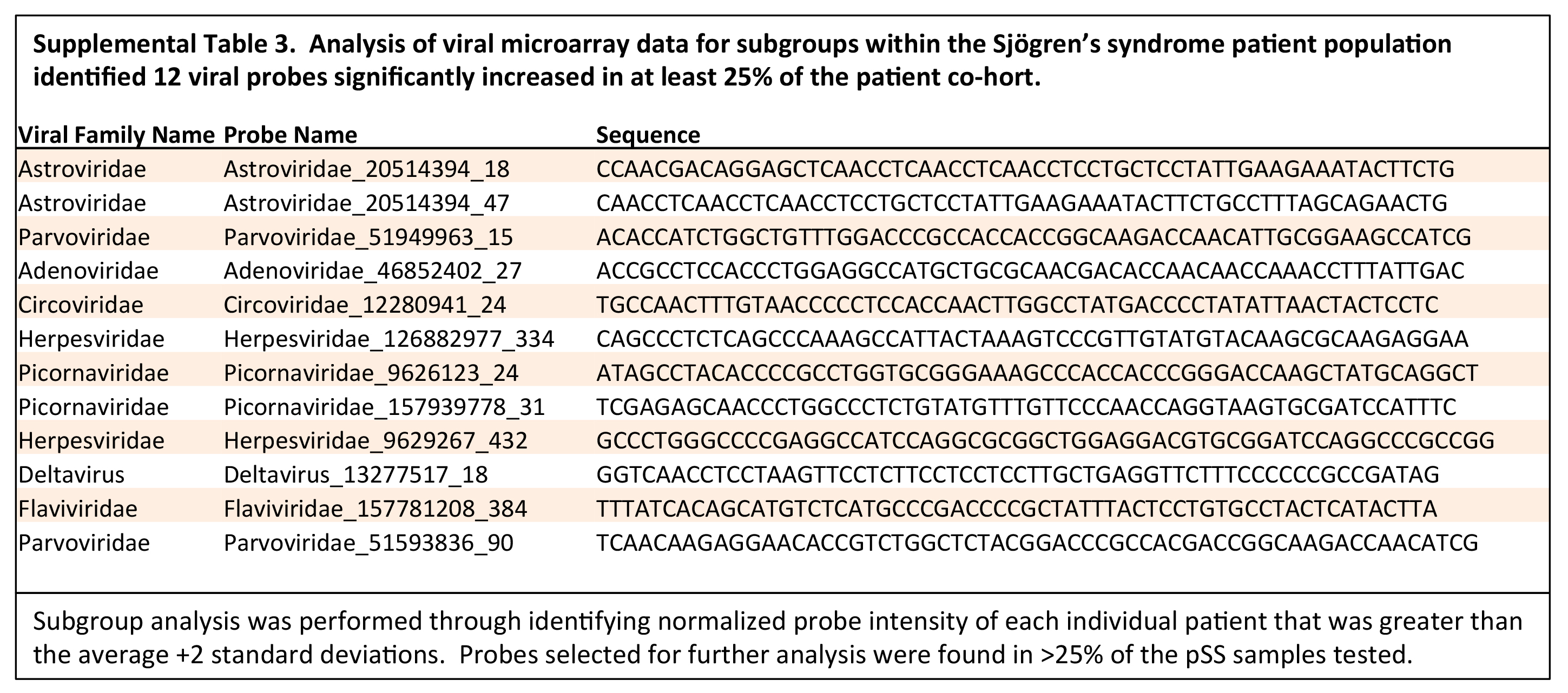
The subgroup analysis of the viral transcripts differentially expressed within subpopulations in the pSS patient cohorts compared to the healthy controls
confirmed the presence of two distinct viral profiles within the pSS patient population. Through the subgroup pSS patient analysis, 12 viral probes were
significantly increased in subpopulations within the pSS patient cohort relative to the healthy controls (Figure 2A, Supplemental Table 3). Significant
pairwise correlation analysis identified 2 distinct viral profiles within the pSS patient population (Figure 2B, Supplemental Table 4). Virus profile I
contained probes recognizing the HDV, Astroviridae, Circoviridae, and Herpesviridae virus families. Virus profile II contained probes recognizing the
Flaviviridae, Picornaviridae, Herpesviridae, and Parvoviridae virus families. Through this subgroup analysis, virus profile I, including HDV, presented the
highest probe intensity and significant increase across the subgroup analysis.
Across both the collective and subgroup analyses, HDV was identified as the most significant virus signature in the pSS cohort present in the affected
salivary gland tissue of Sjögren’s syndrome patients, as defined by significantly elevated HDV probe intensity in the collective and individual pSS
patients relative to healthy controls. Therefore, HDV was selected for further characterization and evaluation as a potential trigger in the development of
a Sjögren’s syndrome phenotype.
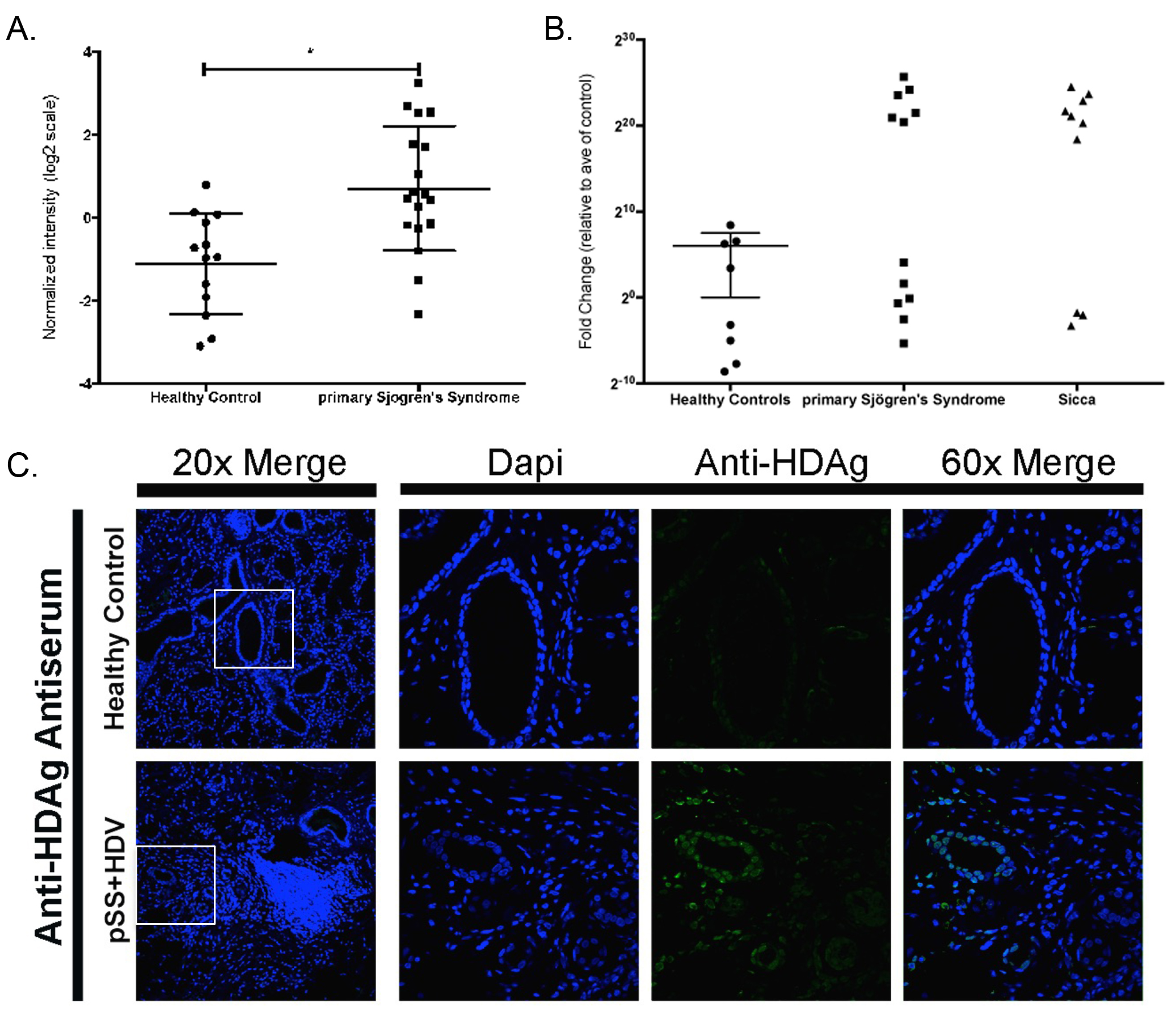
Figure 3. Viral microarray analysis identified HDV in 50% of the patients tested and was confirmed through detection of HDV sequence and antigen in
salivary gland tissue.
(A) Viral microarray analysis rendered a significant increase in HDV in pSS patients relative to healthy controls (P < 0.05, n = 26). (B) qPCR
confirmed the presence of HDV sequence detected in 50% of pSS samples tested. Sicca patients were also positive for the presence of HDV sequence.
Secondary confirmation was performed by an independent lab and detected HDV in 50% of samples tested (Supplemental Figure 1). (C) HDV antigens were
detected in paraffin-embedded salivary gland tissue and rendered a nuclear staining pattern consistent with prior studies of HDV protein localization.
Download as a PowerPoint Slide
Hepatitis Delta Virus Detected in pSS
Microarray analysis identified the presence of hepatitis delta virus (HDV), which was significantly increased in over 50% of the pSS patients compared to
healthy controls (Figure 3A-B). The HDV probe was homologous for the sequence from genotype 1, the genotype most prevalent in North America, Europe, and
the Middle East [34]. Confirmation of HDV viral sequence was performed using nested RT-PCR [25] (Supplemental Figure 1A) using a new RT-qPCR assay targeted to the transcript region of the HDV genome (Figure 3B). To address the potential for
contamination, the hepatitis delta virus sequence detected in minor salivary gland tissue was confirmed in a second pSS cohort in an independent lab with
similar results (Supplemental Figure 1B). Hepatitis delta virus was also detected in a subset of sicca patients experiencing xerostomia and/or
xerophthalmia symptoms but that did not meet the full criteria for pSS diagnosis at the time of tissue biopsy. No data were available at the time of
analysis as to whether patients experiencing sicca symptoms had progressed to develop pSS. Hepatitis delta antigen was detected in HDV-positive pSS in
comparison to salivary gland tissue from healthy controls using confocal immunofluorescence imaging (Figure 3C). Immunohistochemical detection of HDAg
rendered a nuclear localization pattern in HDV-positive paraffin-embedded minor salivary gland tissue. These patterns are similar to those reported for HDV
antigen expression in HDV infected tissues [28
,
35].
Table 1. Clinical parameters of primary Sjögren’s syndrome, primary Sjögren’s syndrome-HOVhigh or primary Sjögren’s syndrome-HDVlow 10w relative to healthy control cohort.
|
|
Healthy Controls
|
Primary Sjögren’s Syndrome
|
Primary Sjögren’s
Syndrome
|
|
|
|
|
HDVlow
|
HDVhigh
|
|
Age
|
42 ± 9
|
50 ± 13
|
54 ± 12#
|
48 ± 14
|
|
Sex (F/M)
|
8/7
|
8/7
|
3/4
|
5/3
|
|
Total Unstimulated Saliva Flow
|
4.77 ± 5.15
|
1.22 ± 1.48*
|
1.04 ± 1.40
|
1.38 ± 1.63
|
|
Total Stimulated Saliva Flow
|
18.28 ± 11.27
|
9.14 ± 6.49*
|
7.56 ± 7.19#
|
10.53 ± 5.94
|
|
Focus Score
|
0.36 ± 0.63
|
1.87 ± 1.25**
|
2.43 ± 1.27 ###
|
1.37 ± 1.06#
|
|
Schirmer’s Test
|
19.2 ± 12.07
|
5.5 ± 4.9**
|
3.8 ± 2.8##
|
7 ± 6.0#
|
|
Anti-Nuclear Antibodies
(% Positive)
|
0%
|
87%***
|
100%###
|
75%##
|
|
Anti-SSA /Ro Antibodies
(% Positive)
|
0%
|
80%***
|
86%###
|
75%###
|
|
Anti-SSB/La Antibodies
(% Positive)
|
0%
|
67%***
|
86%###
|
50%#
|
|
Rheumatoid Factor
(% Positive)
|
17%
|
60%*
|
86%#
|
38%
|
|
lgG (Serum)
|
1146 ± 269
|
1673 ± 578*
|
1859±663##
|
1488±453
|
|
lgM (Serum)
|
137 ± 85
|
104 ± 37
|
95 ± 37
|
113 ± 38
|
|
C3 COMPLEMENT
|
124 ± 22
|
112 ± 29
|
105 ± 23
|
118 ± 35
|
|
C4 COMPLEMENT
|
26 ± 10
|
21 ± 9
|
21 ± 8
|
21 ± 10
|
|
WBC Count
|
5.76 ± 1.44
|
5.09 ± 1.44
|
5.39 ± 1.79
|
4.79 ± 1.02
|
|
Lymphocyte Count
|
30.7 ± 6.2
|
36.0 ± 8.7
|
34.4 ± 8.8
|
37.6 ± 9.0
|
|
Monocyte Count
|
7 ± 3
|
9 ± 1*
|
9 ± 2
|
9 ± 1
|
|
Hematocrit
|
42.1 ± 2.9
|
41.6 ± 1.4
|
41.7 ± 2.4
|
41.4 ± 4.6
|
|
Sed Rate
|
17 ± 9
|
14 ± 8
|
18 ± 9
|
10 ± 5
|
|
Hemoglobin
|
14.1 ± 1.0
|
14.0 ± 1.1
|
14.1 ± 0.7
|
13.8 ± 1.5
|
|
Platelets
|
281 ± 45
|
251 ± 76
|
248 ± 89
|
255 ± 66
|
|
Albumin
|
4.2 ± 0.3
|
4.1 ± 0.3
|
4.0 ± 0.2
|
4.3 ± 0.2☨
|
|
Alkaline Phosphatase (ALP)
|
71 ± 20
|
64 ± 15
|
66 ± 15
|
62 ± 16
|
|
Alanine Transaminase (ALT)
|
24 ± 11
|
27 ± 11
|
24 ± 7
|
30 ± 14
|
|
Amylase
|
72 ± 33
|
92 ± 28
|
100 ± 33
|
83 ± 21
|
|
Aspartate Aminotransferase (AST)
|
24 ± 6
|
31 ± 23
|
26 ± 6
|
37 ± 32
|
|
Calcium
|
2.32 ± 0.09
|
2.39 ± 0.13
|
2.32 ± 0.10
|
2.45 ± 0.13
|
|
Chloride
|
104 ± 3
|
104 ± 1
|
103 ± 1
|
104 ± 1
|
|
Creatinine
|
0.80 ± 0.18
|
0.84 ± 0.16
|
0.83 ± 0.18
|
0.84 ± 0.15
|
|
Glucose
|
98 ± 15
|
92 ± 6
|
92 ± 7
|
91 ± 6
|
|
Magnesium
|
0.90 ± 0.7
|
0.88 ± 0.06
|
0.85 ± 0.5
|
0.90 ± 0.07
|
|
|
|
|
|
|
All values are mean± standard deviation unless otherwise stated. Statistically significant values in primary Sjögren’s Syndrome versus healthy controls *P < 0.05, **P < 0.005, ***P < 0.0005, n = l0-15. Statistically significant values in Primary Sjögren’s Syndrome HDVlow versus Primary Sjögren’s Syndrome HDVhigh ☨ P < 0.05, n = 6-8. Statistically significant values in Primary Sjögren’s Syndrome HDVhigh or Primary Sjögren’s Syndrome HDVlow versus healthy controls #P < 0.05, ##P < 0.005, ###P < 0.0005, n = 6-15. HDVhigh is defined as samples possessing normalized microarray intensity values above 95% Confidence Interval based on healthy controls. HDVlow is defined as samples possessing normalized microarray intensity values within the 95% Confidence Interval based on healthy controls. Statistical analysis was evaluated by t-test or fisher’s exact test for categorical data.
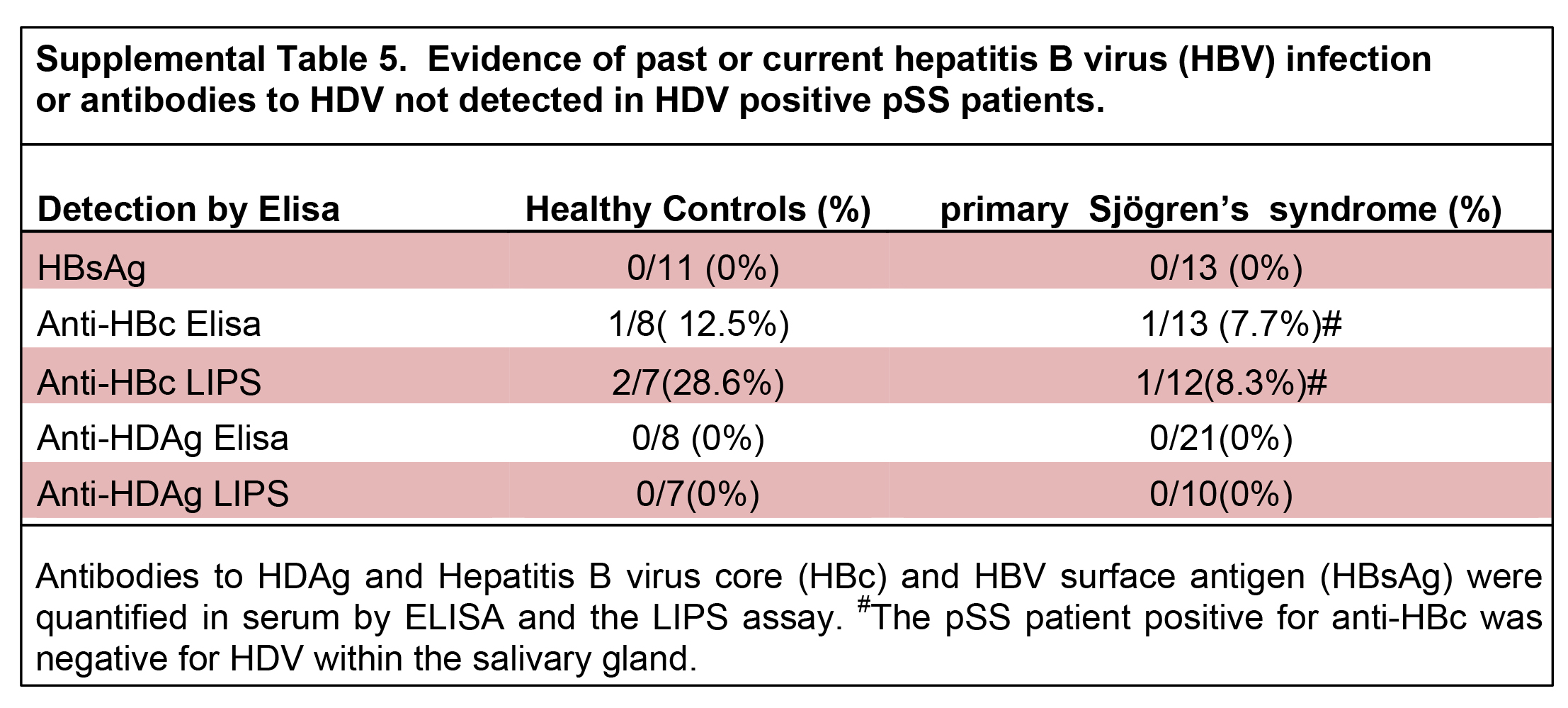
Absence of detectible past or present HBV infection
Primary Sjögren’s syndrome patients positive for HDV in salivary gland tissue lacked evidence of a past or current hepatitis B virus (HBV) coinfection.
Patient serum was tested for HBV surface antigen (HBsAg) and antibodies to HBV core (HBc) (Table 1). All of the patients tested for HBsAg were negative
(Table 1). One healthy control and one pSS serum sample tested were positive for anti-HBc antibodies, but both of the samples positive for anti-HBc
antibodies were negative for HDV sequence in salivary gland tissue (Supplemental Table 5). Luciferase Immunoprecipitation System (LIPS) assay offers a
higher degree of sensitivity in the detection of antibodies [29]. The LIPS assay was also unable to detect anti-HBc antibodies in the serum of pSS patients that were positive for HDV in salivary gland tissue
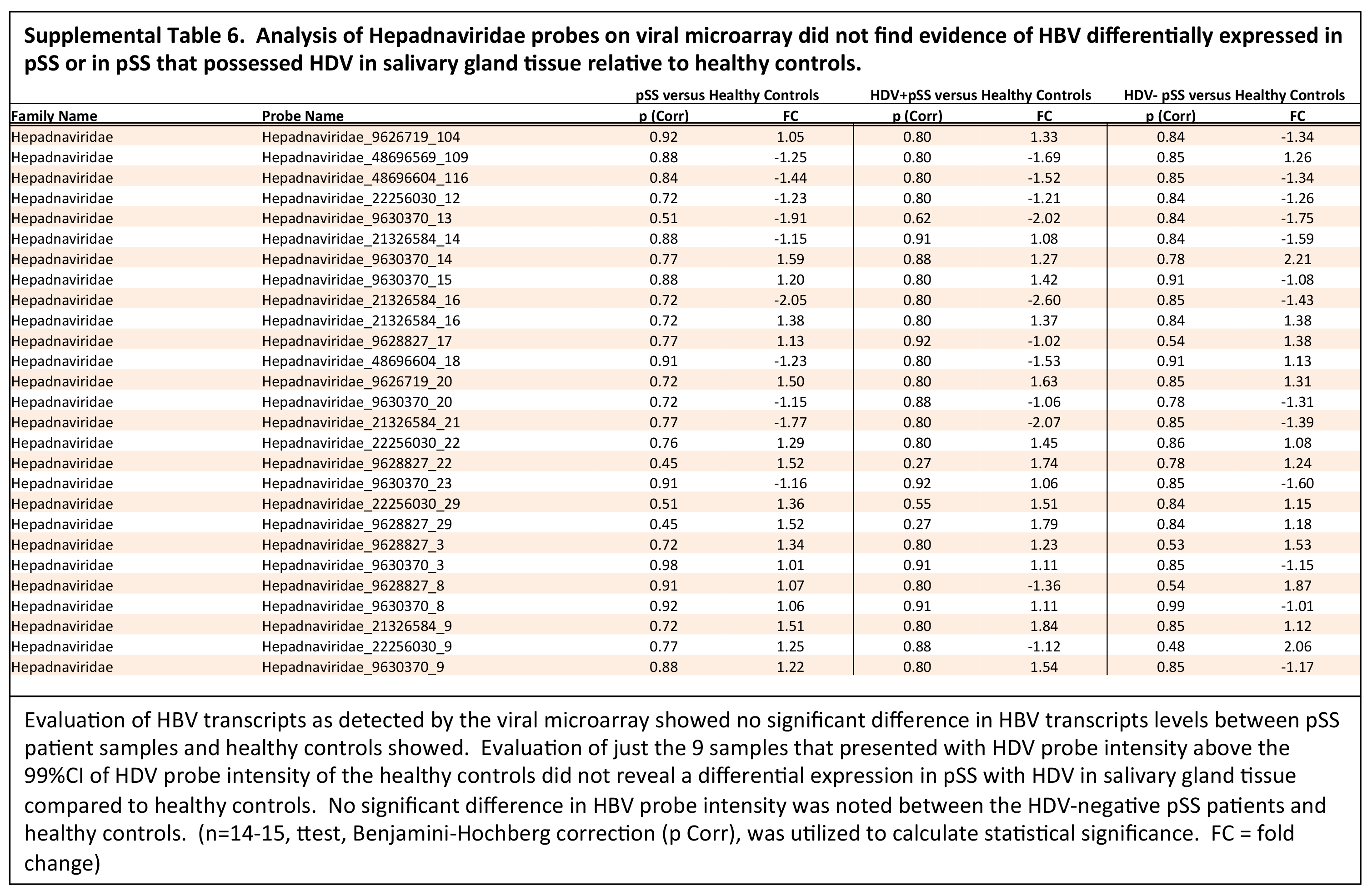
(Supplemental Table 5). These data are in line with reports of similar rates of HBc antibody detection between healthy controls and pSS [36]. Viral microarray contained over 100 probes recognizing hepadnaviridae virus family sequences but lacked significant differential expression between pSS
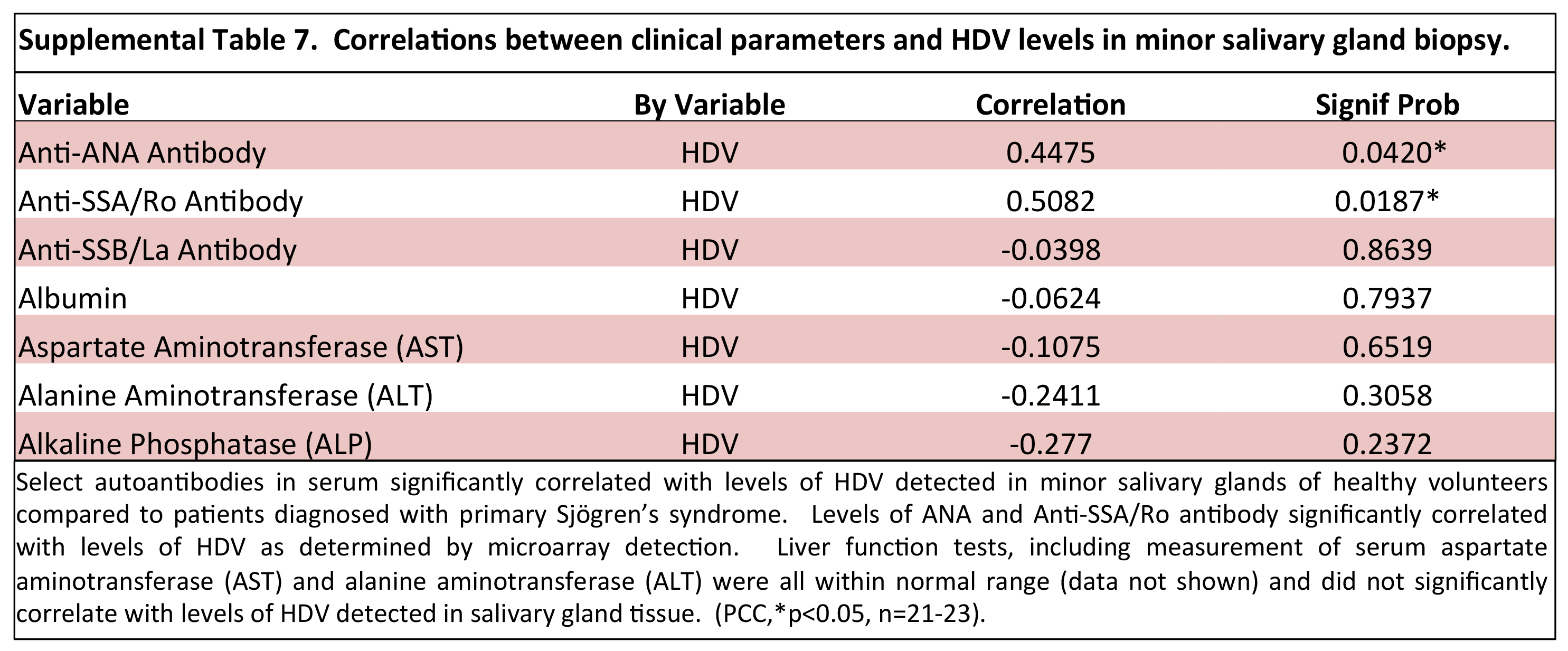
and healthy controls or between HDV-positive pSS and healthy controls (Supplemental Table 6). Transaminase levels were within normal range for the pSS
patient population tested and did not correlate with the level of HDV in salivary gland as detected by viral microarray (Supplemental Table 7). As well,
the patients that tested positive for HDV by microarray lacked detectible antibodies to the HDV antigens (Table 1). In conclusion, pSS patients that tested
positive for HDV in salivary gland tissue were negative for evidence of an active or past HBV infection or a systemic antibody production to HDV antigen.
Clinical Characteristics of HDV in pSS
The pSS patient population was divided into two groups, defined as pSS patients with elevated levels of HDV (positive/HDVhigh, n = 8) and pSS
patients with HDV levels similar to healthy controls (negative/HDVlow, n = 7), as determined by microarray analysis. Analyses were performed to
identify significant changes in clinical parameters between the total pSS population and healthy controls, between HDVhigh and HDVlow
, between HDV-high pSS and healthy controls, and between HDVlow and healthy controls (Table 1).
Analysis of the total pSS patient cohort relative to healthy controls identified significant decreases in unstimulated saliva flow (P < 0.05),
stimulated saliva flow (P < 0.05), and tear production (P < 0.005), and significant increases in focal lymphocytic infiltration (P < 0.005), and levels of total serum IgG and autoantibodies (anti-nuclear, anti-SSA/Ro, anti-SSB/La antibodies, and rheumatoid factor). There was
a limited significant difference identified between the HDVhigh and the HDVlow patient groups. Albumin was elevated in the HDV high subgroup (4.3 ± 0.2, P < 0.05) in comparison to the HDVlow cohort (4.0 ± 0.2). No significant difference was observed in
albumin levels in the total pSS cohort in comparison to healthy controls.
Analysis of clinical parameters in HDVhigh or HDVlow patients relative to healthy controls identified numerous significant
differences in the multiple clinical features analyzed. While there was no significant difference in the age range of the healthy controls (42 ± 9) and total
pSS population (50 ± 13, N.S.), pSS patients in the HDVlow subgroup were significantly older (54 ± 12, P < 0.05) compared to healthy
controls (42 ± 9), in contrast to HDVhigh subgroup (48 ± 14). This study contained a higher percent of males (47%) than normally reported for
Sjögren’s syndrome patient populations (5-10%). No significant difference in sex ratio was observed between the HDVlow and HDVhigh
pSS groups. Total stimulated saliva flow was significantly decreased in the HDVlow pSS group (7.56 ± 7.19, P < 0.05) compared to healthy
controls (18.28 ± 11.27). Focus score was significantly increased in both the HDVlow group (2.43 ± 1.27, P < 0.0001) and HDVhigh
group (1.37 ± 1.06, P < 0.05). Tear production was significantly decreased in the HDVlow group (3.8 ± 2.8, P < 0.005) and HDVhigh group (7.0 ± 6.0, P < 0.05) relative to healthy controls (19.2 ± 12.07) as measured by Schirmer’s test. Together, the HDVhigh patient group displayed a less pronounced clinical presentation of Sjögren’s syndrome symptoms and antibody profiles than the HDV low patient group.
HDV Antigens Induced a pSS-like Phenotype in Vivo
An animal model was developed to evaluate the potential of HDV antigens to initiate development of a Sjögren’s syndrome phenotype in vivo. Hepatitis delta
virus is a negative sense single strand RNA (ssRNA) virus that is classically thought to require a helper virus, hepatitis B virus (HBV), for packaging and
transmission [37]. The ~1700nt circular HDV genome contains a single open reading frame and expresses two proteins, the small antigen (S-HDAg) and the large antigen
(L-HDAg). The S-HDAg is responsible for initiation of RNA genome replication and is expressed early in the infection cycle. The L-HDAg is expressed in
later stages of viral infection and plays a role in ribonucleoprotein (RNP) complex packaging. In the classic HBV:HDV coinfection model, the HDV genome,
S-HDAg, and L-HDAg form the RNP complex and are packaged into the coat membrane of HBV. Due to the absence of HBV in the pSS viral profile, recombinant
adeno-associated virus (rAAV) was used to deliver HDV genes to recapitulate the chronic HDV infection. Female C57BL/6 received rAAV containing S-HDAg,
L-HDAg, or a combination of S-HDAg/L-HDAg expression cassettes through retrograde cannulation to salivary gland tissue. These three in vivo models depicted
different stages of the HDV infection cycle. rAAV containing a luciferase expression cassette was utilized in the control group to account for
rAAV-mediated responses. Immunohistochemical detection of HDAg rendered a nuclear localized pattern of protein expression as delivered by rAAV
(Supplemental Figure 2). Mice were monitored for 4 months post-cannulation for development of autoantibodies, changes in stimulated saliva flow and focal
lymphocytic infiltration in salivary gland tissue.
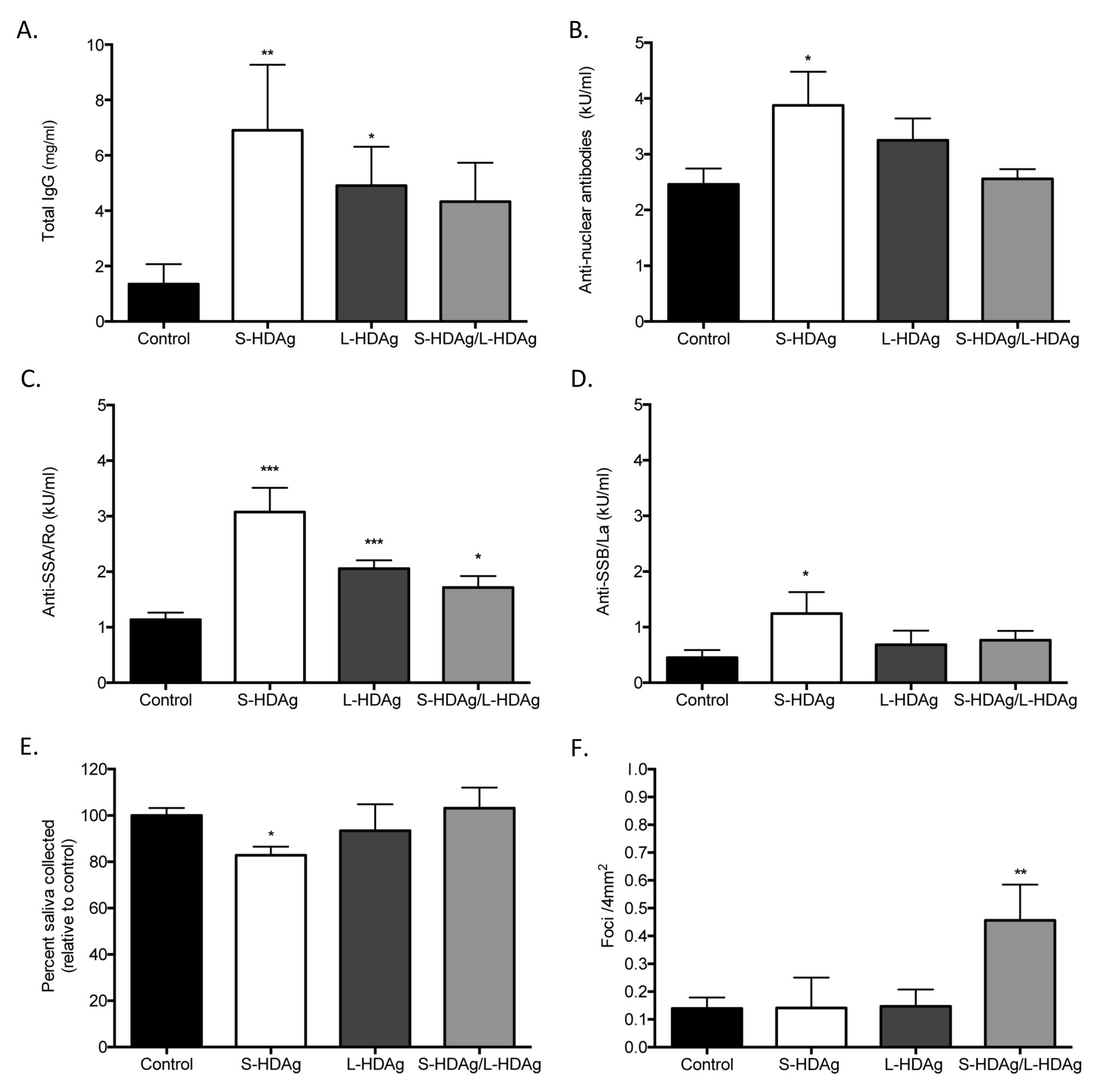
Figure 4. Expression of HDV antigens recapitulates a Sjögren’s syndrome-like phenotype in an in vivomurine model. (
A) Total IgG was significantly increased in mice expressing S-HDAg and L-HDAg, but not in mice that received combination S-HDAg/L-HDAg (t-test, ** P < 0.01, *P < 0.05, n = 7-15). (B) Antinuclear antibodies (ANA) were elevated in mice expressing S-HDAg (t-test, *P < 0.05,
N = 6-11). (C) Anti-SSA/Ro60 antibodies were significantly elevated in mice expressing S-HDAg, L-HDAg, and the combination of S-HDAg/L-HDAg (t-test, ***P < 0.0005, *P < 0.05, n = 7-11). (D) An increase in Anti-SSB/La antibodies was observed in mice that expressed S-HDAg (t-test, * P < 0.05, n = 6-11). (E) Mice were cannulated with adeno-associated virus (AAV) containing expression cassettes for luciferase (control),
S-HDAg, L-HDAg, or a combination of the two HDV antigens (S-HDAg/L-HDAg). Mice that expressed S-HDAg showed a significant decrease in pilocarpine
stimulated saliva flow compared to controls (t-test, *P < 0.05, n = 6-14). (F) A significant increase in foci was noted in mice that expressed a
combination of S-HDAg and L-HDAg (t-test, **P < 0.005, n = 6-14).
Download as a PowerPoint Slide
Expression of HDV antigens in mouse salivary gland tissue triggered the development of autoantibodies (Figure 4A-D). Mice expressing S-HDAg, depicting an
early HDV infection, showed a significant increase in total IgG, ANA, anti-SSA, and anti-SSB levels. Mice expressing L-HDAg showed a significant increase
in total IgG and anti-SSA. Mice expressing a combination of S-HDAg and L-HDAg, depicting a later stage of HDV infection, showed a significant increase in
anti-SSA. The strongest autoantibody profile was observed in mice expressing S-HDAg, representative of early stages of HDV infection.
Expression of S-HDAg in mouse salivary gland tissue resulted in a significant decrease in stimulated saliva flow (Figure 4E). Mice were monitored for
pilocarpine-stimulated saliva flow at 4 months post cannulation. Mice expressing S-HDAg resulted in a statistically significant decrease in
pilocarpine-stimulated saliva flow relative to controls. No change in pilocarpine-stimulated saliva production was noted in mice that expressed the L-HDAg
or combined S-HDAg/L-HDAg expression. Together, our data suggest that the expression of S-HDAg, representative of early stages of HDV infection, possesses
the capacity to impact stimulated saliva flow.
Expression of both S-HDAg and L-HDAg, depicting a later stage of HDV infection, resulted in a significant increase in focal lymphocytic infiltration in
salivary gland tissue. Focal accumulation of lymphocytes within the affected salivary gland is a hallmark in pSS diagnosis. In contrast to the S-HDAg
mediated reduction in saliva flow and strong autoantibody profile, mice that expressed combined S-HDAg/L-HDAg showed a significant increase in lymphocytic
foci (Figure 4F). No significant increase in lymphocytic foci was noted with singular expression of S-HDAg or L-HDAg in the salivary glands compared to
control mice (Figure 4F). The variance in the area of the foci in mice expressing the combination of S-HDAg and L-HDAg was observed to be increased
compared to the area of foci in the control mice (Supplemental Figure 3A-B). Ectopic lymphoid structures were also evident in mice expressing combined
S-HDAg/L-HDAg (Supplemental Figure 3). Additionally, similar to the pSS patient cohort, mice expressing HDV antigens in salivary gland tissue did not
develop detectible antibodies to HDV antigens (data not shown).
Discussion
This study identified the presence of HDV in a subset of Sjögren’s syndrome patients. The detection of HDV by custom viral microarray was subsequently
confirmed by detection of the HDV antigen in affected salivary gland tissue and detection of the HDV sequence in two pSS cohorts and by an independent research
team. Animal models were developed to evaluate the potential for HDV antigens expressed in salivary gland tissue to trigger development of a Sjögren’s
syndrome-like phenotype. We have shown that the expression of HDV antigens in mouse salivary gland recapitulates the development of a complete pSS-like
phenotype in vivo. Together, the identification of HDV in the affected salivary gland tissue of Sjögren’s syndrome patients and the demonstrated ability of
HDV antigens to trigger the pathogenesis of a pSS-like phenotype in vivo provide further support for a viral-mediated mechanism in the development of
Sjögren’s syndrome.
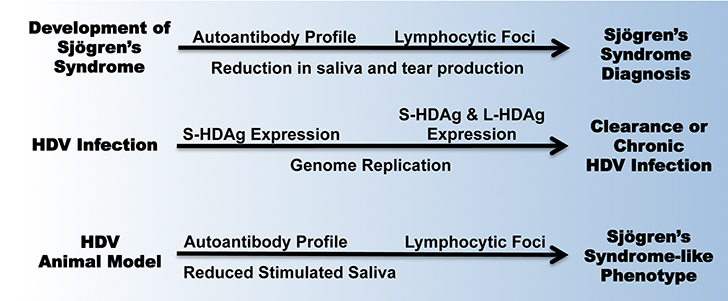
Figure 5. Progression of Sjögren’s syndrome disease mirrors the development of HDV infection and HDV antigen-mediated induction of a Sjögren’s
syndrome-like phenotype in vivo.
Download as a PowerPoint Slide
The timeline for developing Sjögren’s syndrome pathogenesis mirrors the HDV-mediated Sjögren’s syndrome phenotype observed in our animal model and in the
known HDV infection cycle (Figure 5). Sjögren’s syndrome patients have reported subjective xerostomia and develop an autoantibody profile years prior to
disease diagnosis [12-14]. Mirroring this behavior, the mice expressing S-HDAg, representing an early stage of HDV infection, presented the strongest autoantibody profile and
significant reduction in stimulated saliva flow. In humans, lymphocytic infiltrates in the salivary gland tissue are evaluated and detected at later stages
of pSS pathogenesis. In the mouse model, the combined expression of S-HDAg and L-HDAg, depicting a later stage HDV infection profile, resulted in a
significant accumulation of lymphocytic foci in vivo. Together, the chronological development of Sjögren’s syndrome echoes the progression of a chronic,
low-grade HDV infection in the salivary gland as observed in our HDV animal model.
Chronic HDV infections have previously been associated with triggering a clear antiviral response and development of autoimmunity. Multiple studies have
shown the capacity of HDV antigens to stimulate expression of or directly interact with multiple interferon-stimulated genes and genes associated with pSS
pathogenesis, including SSB/La protein [38-41]. Prior studies have associated HDV infections with the development of autoantibodies, including ANA, liver-kidney microsomal antibodies (LKM), and smooth
muscle antibodies (SMA), among others [42-44]. Our data, in connection with prior reports of HDV-mediated autoantibody development, further support the potential of HDV to trigger development of
pSS-associated autoimmunity. The type of autoantibody profile developed in the presence of HDV may be dependent on the tissues and cellular localization of
active HDV persistence.
Detection of HDV in the salivary gland tissue of Sjögren’s syndrome patients in the absence of HBV points to a unique tissue tropism and/or mechanism of
persistence not previously identified. Hepatitis delta virus can readily replicate its genome and antigens using host cellular mechanisms, enabling
persistence in the absence of an active helper virus infection, and has been shown to persist in tissue culture at low levels for at least a year in the
absence of a helper virus [27,
37,
38]. Therefore, once inside the cell, HDV is able to establish a chronic presence. In our hands, evidence of a past or current HBV infection has not been
identified in affected salivary gland tissue in pSS patients that are HDV positive. This is in line with prior studies evaluating the incidence of HBV
infection in pSS studies that have found similar or lower HBV infection rates in pSS patients [36, 45, 46]. The question remains as to how HDV gains entry into the salivary glands of pSS patients in the absence of a detectible current or past HBV infection.
Prior studies have shown cell-mediated immune response to low-level viral infections and/or exposures in the absence of antibody production [47]. Further studies are needed to evaluate the potential of cell-mediated immune response to low level HDV and HBV antigens in the absence of a detectible
humoral immune response in both the pSS patient population and HDV animal models.
Further analysis is needed to identify primary routes of infection and viable antiviral therapeutics. Prior studies have shown the sequence homology of
genotype 1 HDV infections associated with HDV:HBV coinfection ranges between 91.2% and 91.7% [48
,
49]. In our study, a higher percent of sequence homology of 98.5% (between 97.3%-99.3%) was observed in pSS patients. This high degree of sequence homology
between patient samples may be a factor of the low level rate of genome replication or a stable primary reservoir. Multiple therapies are being evaluated
for treatment of chronic HDV infection associated with HBV coinfection, including lonafarnib, tipifarnib, and FTI-277 [50, 51]. The primary mechanism of these drugs inhibits farnesyl transferase activity, which is required for the post-translational modification of the L-HDAg
facilitating interaction with HBsAg and packaging in the HBV virion. While these drugs have shown success in treating HDV in relation to HDV:HBV
coinfection, it is unclear at this stage whether these drugs would have a similar effect in the chronic HDV present in salivary gland tissues of Sjögren’s
syndrome patients and in the absence of an active HBV infection. Ongoing studies are focused on how HDV gains entry into salivary gland tissue in the
absence of a detectible HBV infection and the underlying mechanism(s) of HDV-mediated pSS disease pathogenesis.
Multiple viral infections have been associated with the development of sialadenitis and sicca symptoms similar to that in pSS patients. As primary
examples, hepatitis C virus (HCV) and human immunodeficiency virus (HIV) infections are noted to trigger sialadenitis, sicca symptoms, and distinct
autoantibody profiles [52-54]. Identifying the association between these viral infections and Sjögren’s syndrome symptoms has refocused the therapeutic approach to treating the
underlying viral infection. HIV and HCV are now exclusion criteria in the diagnosis of Sjögren’s syndrome [11]. The identification of HDV in pSS patients and the recapitulation of disease phenotype in vivo follows a similar path of study previously reported for
HCV in pSS populations and in vivo [52, 55]. Therefore, we postulate that HDV may be another viral infection resulting in the development of a Sjögren’s syndrome-like phenotype, and further
analysis is warranted to evaluate the diagnosis of a chronic HDV infection as an exclusion criterion for the diagnosis of Sjögren’s syndrome. Identifying
the Sjögren’s syndrome patient population that is HDV positive will refocus the therapeutic approach from generalized immune suppression to targeted
antiviral therapies.
Prior studies that have introduced the expression of viral proteins, including HCV and HTLV-1 proteins, or endogenous proteins associated with pSS,
including bone morphogenic protein 6 (BMP-6), have shown incomplete primary Sjögren’s syndrome-like phenotypes [24, 55, 56]. Most notably, these prior studies have lacked the production of pSS-associated autoantibodies to SSA/Ro or SSB/La proteins. Our study identified the
increased presence of HDV in the salivary gland tissue of pSS patients and targeted expression of HDV antigens in murine salivary glands resulted in the
development of a complete Sjögren’s syndrome phenotype. Together, these data suggest that the simple expression of a viral or endogenous protein or general
viral infections in the salivary gland are not sufficient to elicit a complete pSS-like disease phenotype and that the pSS disease pathogenesis may be a
targeted response to a specific viral exposure.
Analysis of clinical features based on the level of HDV within affected salivary gland tissue revealed a less pronounced disease symptomology in the HDV high pSS patients. The detection of HDV in patients experiencing sicca symptoms but not meeting full criteria for pSS diagnosis may provide an
early window for diagnosis and intervention prior to progression of pSS-associated pathology. Pertovaara et al. reported that 36% of patients
experiencing sicca symptoms progress to development of pSS [57]. Our current hypothesis is that HDV-positive sicca patients may either be at early stages of the disease progression or are missing a cellular or genetic
susceptibility factor required for progression of disease pathology. Further studies are needed to clarify the clinical differences between HDV high and HDVlow pSS patients and to evaluate disease progression or resolution in HDV-positive sicca patients to help define the
susceptibility factors supporting or restricting chronic persistence of HDV in salivary gland tissue.
Primary Sjögren’s syndrome is not considered to be an infectious or contagious disease. Therefore multiple factors, including genetic susceptibility,
favorable cellular and/or tissue environment, and/or exposure to pathogen(s), may be required to promote a viral-mediated development of pSS pathology.
Further large-scale studies are needed to define the incidence of HDV in global pSS patient cohorts and the underlying genetic susceptibility factors
impacting HDV-mediated disease pathogenesis.
Financial Support
The funding for this study was provided through the National Institute of Dental and Craniofacial Research, the National Institutes of Health K99/R00
Pathway to Independence Award (K99 DE21745), and National Institute of Dental and Craniofacial Research, National Institutes of Health (1ZIADE000695).
References
1. Knobler SL, O’Connor S, Lemon SM, Najafi M, eds. The Infectious Etiology of Chronic Diseases: Defining the Relationship, Enhancing the Research, and
Mitigating the Effects: Workshop Summary. The National Academies Collection: Reports funded by National Institutes of Health. Washington (DC)2004.
2. Igoe A, Scofield RH. Autoimmunity and infection in Sjögren’s syndrome. Current opinion in rheumatology. 2013 Jul;25(4):480-7. PubMed PMID: 23719365.
doi: 10.1097/BOR.0b013e32836200d2
3. Meyer O. Interferons and autoimmune disorders. Joint, bone, spine: revue du rhumatisme. 2009 Oct;76(5):464-73. PubMed PMID: 19773191. doi: 10.1016/j.jbspin.2009.03.012
4. Nandula SR, Dey P, Corbin KL, Nunemaker CS, Bagavant H, Deshmukh US. Salivary gland hypofunction induced by activation of innate immunity is
dependent on type I interferon signaling. Journal of oral pathology & medicine: official publication of the International Association of Oral
Pathologists and the American Academy of Oral Pathology. 2013 Jan;42(1):66-72. PubMed PMID: 22672212. Pubmed Central PMCID: 3443546.
doi: 10.1111/j.1600-0714.2012.01181.x
5. Nguyen CQ, Peck AB. The Interferon-Signature of Sjögren’s Syndrome: How Unique Biomarkers Can Identify Underlying Inflammatory and
Immunopathological Mechanisms of Specific Diseases. Frontiers in immunology. 2013;4:142. PubMed PMID: 23847613. Pubmed Central PMCID: 3701867. doi: 10.3389/fimmu.2013.00142
6. Ronnblom L, Eloranta ML. The interferon signature in autoimmune diseases. Current opinion in rheumatology. 2013 Mar;25(2):248-53. PubMed PMID:
23249830. doi: 10.1097/BOR.0b013e32835c7e32
7. Vakaloglou KM, Mavragani CP. Activation of the type I interferon pathway in primary Sjögren’s syndrome: an update. Current opinion in rheumatology.
2011 Sep;23(5):459-64. PubMed PMID: 21760509. doi: 10.1097/BOR.0b013e328349fd30
8. Yao Y, Liu Z, Jallal B, Shen N, Ronnblom L. Type I interferons in Sjögren’s syndrome. Autoimmunity reviews. 2013 Mar;12(5):558-66. PubMed PMID:
23201923. doi: 10.1016/j.autrev.2012.10.006
9. Binard A, Devauchelle-Pensec V, Fautrel B, Jousse S, Youinou P, Saraux A. Epidemiology of Sjögren’s syndrome: where are we now? Clinical and
experimental rheumatology. 2007 Jan-Feb;25(1):1-4. PubMed PMID: 17417982.
10. Bureau USC. U.S. and World Population Clock 2013 [cited 2013 12/20/2013]. 7131957000 at 3pm on 12/20/13; M. Weller]. Available from:
http://www.census.gov/popclock/
11. Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, Daniels TE, Fox PC, Fox RI, Kassan SS, Pillemer SR, Talal N, Weisman
MH. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group.
Annals of the rheumatic diseases. 2002 Jun;61(6):554-8. PubMed PMID: 12006334. Pubmed Central PMCID: 1754137.
12. Jonsson R, Theander E, Sjöstrom B, Brokstad K, Henriksson G. Autoantibodies present before symptom onset in primary Sjögren syndrome. JAMA: the
journal of the American Medical Association. 2013 Nov 6;310(17):1854-5. PubMed PMID: 24193084. doi: 10.1001/jama.2013.278448
13. Al-Hashimi I, Khuder S, Haghighat N, Zipp M. Frequency and predictive value of the clinical manifestations in Sjögren’s syndrome. Journal of oral
pathology & medicine: official publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology. 2001
Jan;30(1):1-6. PubMed PMID: 11140894.
14. Mignogna MD, Fedele S, Lo Russo L, Lo Muzio L, Wolff A. Sjögren’s syndrome: the diagnostic potential of early oral manifestations preceding
hyposalivation/xerostomia. Journal of oral pathology & medicine: official publication of the International Association of Oral Pathologists and the
American Academy of Oral Pathology. 2005 Jan;34(1):1-6. PubMed PMID: 15610399. doi: 10.1111/j.1600-0714.2004.00264.x
15. Ramos-Casals M, Solans R, Rosas J, Camps MT, Gil A, Del Pino-Montes J, Calvo-Alen J, Jimenez-Alonso J, Mico ML, Beltran J, Belenguer R, Pallares L.
Primary Sjögren syndrome in Spain: clinical and immunologic expression in 1010 patients. Medicine. 2008 Jul;87(4):210-9. PubMed PMID: 18626304. doi: 10.1097/MD.0b013e318181e6af
16. Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, Lazure T, Jacques S, Ba N, Ittah M, Lepajolec C, Labetoulle M, Ardizzone M, Sibilia
J, Fournier C, Chiocchia G, Mariette X. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjögren’s
syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2006 Feb 21;103(8):2770-5. PubMed PMID: 16477017. Pubmed
Central PMCID: 1413808. doi: 10.1073/pnas.0510837103
17. Li H, Ice JA, Lessard CJ, Sivils KL. Interferons in Sjögren’s Syndrome: Genes, Mechanisms, and Effects. Frontiers in immunology. 2013;4:290. PubMed
PMID: 24062752. Pubmed Central PMCID: 3778845. doi: 10.3389/fimmu.2013.00290
18. Hall JC, Baer AN, Shah AA, Criswell LA, Shiboski CH, Rosen A, Casciola-Rosen L. Molecular subsetting of interferon pathways in Sjögren’s syndrome.
Arthritis Rheumatol. 2015 May 18. PubMed PMID: 25988820. doi: 10.1002/art.39204
19. Toussirot E, Roudier J. Epstein-Barr virus in autoimmune diseases. Best practice & research Clinical rheumatology. 2008 Oct;22(5):883-96.
PubMed PMID: 19028369. doi: 10.1016/j.berh.2008.09.007
20. Triantafyllopoulou A, Tapinos N, Moutsopoulos HM. Evidence for coxsackievirus infection in primary Sjögren’s syndrome. Arthritis and rheumatism.
2004 Sep;50(9):2897-902. PubMed PMID: 15457458. doi: 10.1002/art.20463
21. Gottenberg JE, Pallier C, Ittah M, Lavie F, Miceli-Richard C, Sellam J, Nordmann P, Cagnard N, Sibilia J, Mariette X. Failure to confirm
coxsackievirus infection in primary Sjögren’s syndrome. Arthritis and rheumatism. 2006 Jun;54(6):2026-8. PubMed PMID: 16732567. doi:
10.1002/art.21906
22. Lee SJ, Lee JS, Shin MG, Tanaka Y, Park DJ, Kim TJ, Park YW, Lee SS. Detection of HTLV-1 in the labial salivary glands of patients with Sjögren’s
syndrome: a distinct clinical subgroup? The Journal of rheumatology. 2012 Apr;39(4):809-15. PubMed PMID: 22382336. doi:
10.3899/jrheum.111075
23. Wang D, Coscoy L, Zylberberg M, Avila PC, Boushey HA, Ganem D, DeRisi JL. Microarray-based detection and genotyping of viral pathogens. Proc Natl
Acad Sci U S A. 2002 Nov 26;99(24):15687-92. PubMed PMID: 12429852. Pubmed Central PMCID: PMC137777. doi: 10.1073/pnas.242579699
24. Yin H, Cabrera-Perez J, Lai Z, Michael D, Weller M, Swaim WD, Liu X, Catalan MA, Rocha EM, Ismail N, Afione S, Rana NA, Di Pasquale G, Alevizos I,
Ambudkar I, Illei GG, Chiorini JA. Association of bone morphogenetic protein 6 with exocrine gland dysfunction in patients with Sjögren’s syndrome and
in mice. Arthritis and rheumatism. 2013 Dec;65(12):3228-38. PubMed PMID: 23982860. doi: 10.1002/art.38123
25. Smedile A, Niro MG, Rizzetto M. Detection of serum HDV RNA by RT-PCR. Methods in molecular medicine. 2004;95:85-93. PubMed PMID: 14982056. doi: 10.1385/1-59259-669-X:85
26. Biosystems A. April 2014. Available from:
http://www3.appliedbiosystems.com/cms/groups/mcb_support/documents/generaldocuments/cms_042380.pdf
27. Chang J, Gudima SO, Tarn C, Nie X, Taylor JM. Development of a novel system to study hepatitis delta virus genome replication. Journal of virology.
2005 Jul;79(13):8182-8. PubMed PMID: 15956563. Pubmed Central PMCID: 1143748. doi: 10.1128/JVI.79.13.8182-8188.2005
28. Bichko VV, Taylor JM. Redistribution of the delta antigens in cells replicating the genome of hepatitis delta virus. J Virol. 1996
Nov;70(11):8064-70. PubMed PMID: 8892931. Pubmed Central PMCID: PMC190880.
29. Burbelo PD, Ching KH, Klimavicz CM, Iadarola MJ. Antibody Profiling by Luciferase Immunoprecipitation Systems (LIPS). J Vis Exp. 2009 (32). PubMed
PMID: 19812534. doi: 10.3791/1549
30. Burbelo PD, Ching KH, Mattson TL, Light JS, Bishop LR, Kovacs JA. Rapid antibody quantification and generation of whole proteome antibody response
profiles using LIPS (luciferase immunoprecipitation systems). Biochemical and biophysical research communications. 2007 Jan 26;352(4):889-95. PubMed
PMID: 17157815. doi: 10.1016/j.bbrc.2006.11.140
31. Zheng C, Voutetakis A, Goldstein B, Afione S, Rivera VM, Clackson T, Wenk ML, Boyle M, Nyska A, Chiorini JA, Vallant M, Irwin RD, Baum BJ.
Assessment of the safety and biodistribution of a regulated AAV2 gene transfer vector after delivery to murine submandibular glands. Toxicological
sciences: an official journal of the Society of Toxicology. 2011 Sep;123(1):247-55. PubMed PMID: 21625005. Pubmed Central PMCID: 3164441. doi: 10.1093/toxsci/kfr144
32. Weller ML, Amornphimoltham P, Schmidt M, Wilson PA, Gutkind JS, Chiorini JA. Epidermal growth factor receptor is a co-receptor for adeno-associated
virus serotype 6. Nature medicine. 2010 Jun;16(6):662-4. PubMed PMID: 20473307. Pubmed Central PMCID: 2885716. doi: 10.1038/nm.2145
33. Wong CW, Heng CL, Wan Yee L, Soh SW, Kartasasmita CB, Simoes EA, Hibberd ML, Sung WK, Miller LD. Optimization and clinical validation of a pathogen
detection microarray. Genome biology. 2007;8(5):R93. PubMed PMID: 17531104. Pubmed Central PMCID: 1929155. doi:
10.1186/gb-2007-8-5-r93
34. Wedemeyer H, Manns MP. Epidemiology, pathogenesis and management of hepatitis D: update and challenges ahead. Nature reviews Gastroenterology &
hepatology. 2010 Jan;7(1):31-40. PubMed PMID: 20051970. doi: 10.1038/nrgastro.2009.205
35. Netter HJ, Kajino K, Taylor JM. Experimental transmission of human hepatitis delta virus to the laboratory mouse. J Virol. 1993 Jun;67(6):3357-62.
PubMed PMID: 8497056. Pubmed Central PMCID: PMC237679.
36. Ram M, Anaya JM, Barzilai O, Izhaky D, Porat Katz BS, Blank M, Shoenfeld Y. The putative protective role of hepatitis B virus (HBV) infection from
autoimmune disorders. Autoimmunity reviews. 2008 Sep;7(8):621-5. PubMed PMID: 18603025. doi: 10.1016/j.autrev.2008.06.008
37. Casey JL. Hepatitis delta virus. Berlin; New York: Springer; 2006. vii, 228 p.
38. Giersch K, Helbig M, Volz T, Allweiss L, Mancke LV, Lohse AW, Polywka S, Pollok JM, Petersen J, Taylor J, Dandri M, Lutgehetmann M. Persistent
hepatitis D virus mono-infection in humanized mice is efficiently converted by hepatitis B virus to a productive co-infection. Journal of hepatology.
2014 Mar;60(3):538-44. PubMed PMID: 24280293. doi: 10.1016/j.jhep.2013.11.010
39. Greco-Stewart V, Pelchat M. Interaction of host cellular proteins with components of the hepatitis delta virus. Viruses. 2010 Jan;2(1):189-212.
PubMed PMID: 21994607. Pubmed Central PMCID: PMC3185554. doi: 10.3390/v2010189
40. Mota S, Mendes M, Penque D, Coelho AV, Cunha C. Changes in the proteome of Huh7 cells induced by transient expression of hepatitis D virus RNA and
antigens. J Proteomics. 2008 Apr 30;71(1):71-9. PubMed PMID: 18541475. doi: 10.1016/j.jprot.2007.12.002
41. Giersch K, Allweiss L, Volz T, Helbig M, Bierwolf J, Lohse AW, Pollok JM, Petersen J, Dandri M, Lutgehetmann M. Hepatitis Delta co-infection in
humanized mice leads to pronounced induction of innate immune responses in comparison to HBV mono-infection. Journal of hepatology. 2015 Mar 17. PubMed
PMID: 25795587. doi: 10.1016/j.jhep.2015.03.011
42. Amengual MJ, Catalfamo M, Pujol A, Juarez C, Gelpi C, Rodriguez JL. Autoantibodies in chronic delta virus infection recognize a common protein of
46 kD in rat forestomach basal cell layer and stellate thymic epithelial cells. Clinical and experimental immunology. 1989 Oct;78(1):80-4. PubMed PMID:
2680184. Pubmed Central PMCID: 1534589.
43. Philipp T, Obermayer-Straub P, Manns MP. Autoantibodies in hepatitis delta. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie.
1995;49(7-8):344-9. PubMed PMID: 8562861.
44. Zauli D, Crespi C, Bianchi FB, Craxi A, Pisi E. Autoimmunity in chronic liver disease caused by hepatitis delta virus. Journal of clinical
pathology. 1986 Aug;39(8):897-9. PubMed PMID: 3745480. Pubmed Central PMCID: 500125.
45. Marcos M, Alvarez F, Brito-Zeron P, Bove A, Perez-De-Lis M, Diaz-Lagares C, Sanchez-Tapias JM, Ramos-Casals M. Chronic hepatitis B virus infection
in Sjögren’s syndrome. Prevalence and clinical significance in 603 patients. Autoimmunity reviews. 2009 Jun;8(7):616-20. PubMed PMID: 19393197. doi: 10.1016/j.autrev.2009.02.007
46. Chen MH, Hsiao LT, Tsai CY, Huang YH, Chou CT. Clinical significance of chronic hepatitis B virus infection in patients with primary Sjögren’s
syndrome. Clinical rheumatology. 2012 Feb;31(2):309-15. PubMed PMID: 21809004. doi: 10.1007/s10067-011-1814-2
47. El-Kamary SS, Hashem M, Saleh DA, Abdelwahab SF, Sobhy M, Shebl FM, Shardell MD, Strickland GT, Shata MT. Hepatitis C virus-specific cell-mediated
immune responses in children born to mothers infected with hepatitis C virus. J Pediatr. 2013 Jan;162(1):148-54. PubMed PMID: 22883419. Pubmed Central
PMCID: PMC3526784. doi: 10.1016/j.jpeds.2012.06.057
48. Niro GA, Smedile A, Andriulli A, Rizzetto M, Gerin JL, Casey JL. The predominance of hepatitis delta virus genotype I among chronically infected
Italian patients. Hepatology. 1997 Mar;25(3):728-34. PubMed PMID: 9049226. doi: 10.1002/hep.510250339
49. Shakil AO, Hadziyannis S, Hoofnagle JH, Di Bisceglie AM, Gerin JL, Casey JL. Geographic distribution and genetic variability of hepatitis delta
virus genotype I. Virology. 1997 Jul 21;234(1):160-7. PubMed PMID: 9234957. doi: 10.1006/viro.1997.8644
50. Koh C, Canini L, Dahari H, Zhao X, Uprichard SL, Haynes-Williams V, Winters MA, Subramanya G, Cooper SL, Pinto P, Wolff EF, Bishop R, Ai Thanda Han
M, Cotler SJ, Kleiner DE, Keskin O, Idilman R, Yurdaydin C, Glenn JS, Heller T. Oral prenylation inhibition with lonafarnib in chronic hepatitis D
infection: a proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. Lancet Infect Dis. 2015 Jul 16. PubMed PMID: 26189433. doi:
10.1016/S1473-3099(15)00074-2
51. Bordier BB, Ohkanda J, Liu P, Lee SY, Salazar FH, Marion PL, Ohashi K, Meuse L, Kay MA, Casey JL, Sebti SM, Hamilton AD, Glenn JS. In vivo
antiviral efficacy of prenylation inhibitors against hepatitis delta virus. J Clin Invest. 2003 Aug;112(3):407-14. PubMed PMID: 12897208. Pubmed
Central PMCID: PMC166292. doi: 10.1172/jci17704
52. Ramos-Casals M, Loustaud-Ratti V, De Vita S, Zeher M, Bosch JA, Toussirot E, Medina F, Rosas J, Anaya JM, Font J, Group S-HS. Sjögren syndrome
associated with hepatitis C virus: a multicenter analysis of 137 cases. Medicine. 2005 Mar;84(2):81-9. PubMed PMID: 15758837.
53. Schiodt M, Greenspan D, Daniels TE, Nelson J, Leggott PJ, Wara DW, Greenspan JS. Parotid gland enlargement and xerostomia associated with labial
sialadenitis in HIV-infected patients. J Autoimmun. 1989 Aug;2(4):415-25. PubMed PMID: 2789646.
54. Zandman-Goddard G, Shoenfeld Y. HIV and autoimmunity. Autoimmun Rev. 2002 Dec;1(6):329-37. PubMed PMID: 12848988.
55. Koike K, Moriya K, Ishibashi K, Yotsuyanagi H, Shintani Y, Fujie H, Kurokawa K, Matsuura Y, Miyamura T. Sialadenitis histologically resembling
Sjögren syndrome in mice transgenic for hepatitis C virus envelope genes. Proc Natl Acad Sci U S A. 1997 Jan 7;94(1):233-6. PubMed PMID: 8990191.
Pubmed Central PMCID: PMC19297.
56. Green JE, Hinrichs SH, Vogel J, Jay G. Exocrinopathy resembling Sjögren’s syndrome in HTLV-1 tax transgenic mice. Nature. 1989 Sep
7;341(6237):72-4. PubMed PMID: 2788824. doi: 10.1038/341072a0
57. Pertovaara M, Korpela M, Uusitalo H, Pukander J, Miettinen A, Helin H, Pasternack A. Clinical follow up study of 87 patients with sicca symptoms
(dryness of eyes or mouth, or both). Annals of the rheumatic diseases. 1999 Jul;58(7):423-7. PubMed PMID: 10381486. Pubmed Central PMCID: 1752915.
Supplementary Files

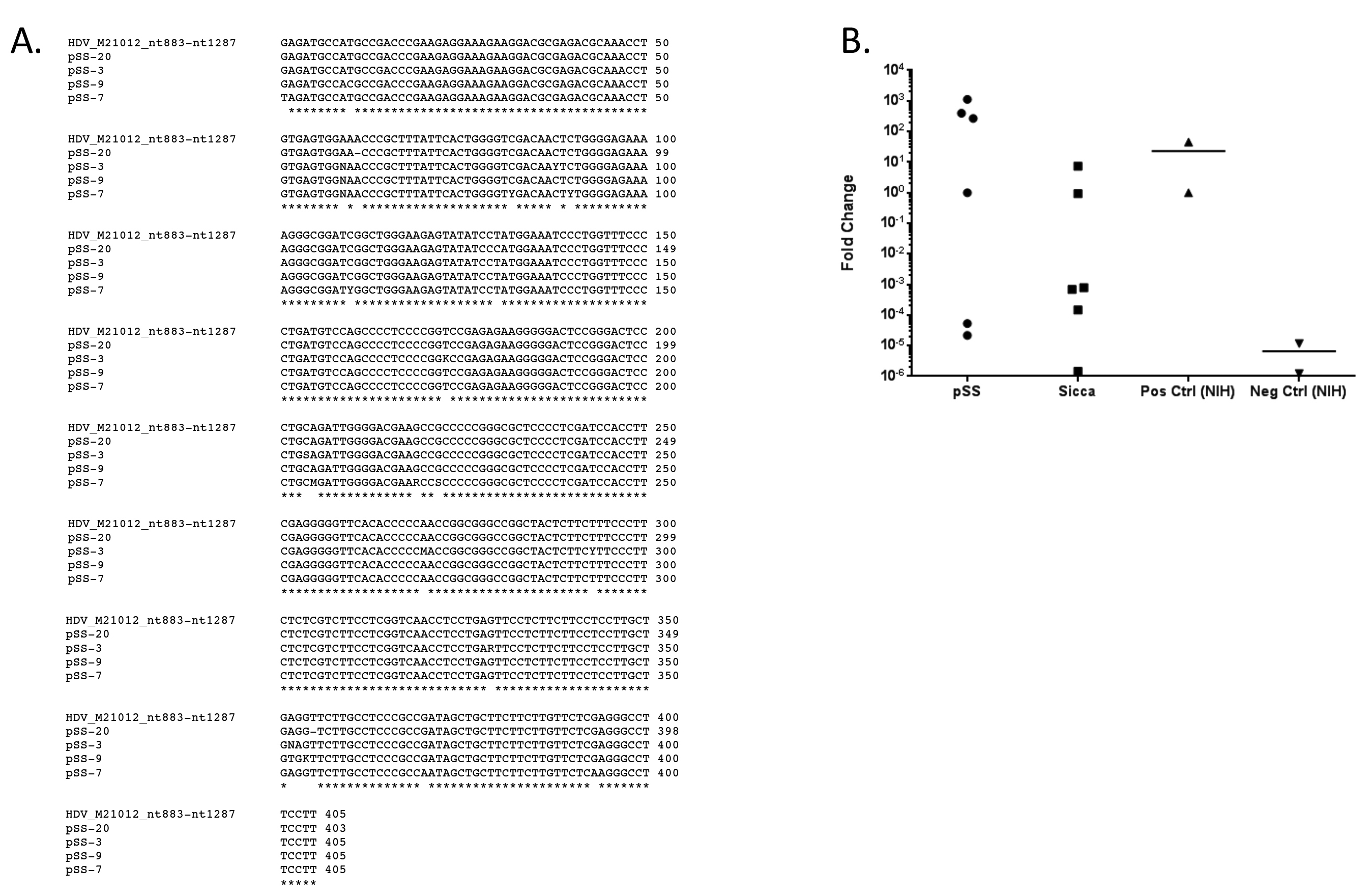
Supplemental Figure 1. HDV sequence was detected in salivary glands obtained from pSS and sicca patients. A) Nested
PCR was used to detect HDV genomic sequence in minor salivary gland tissue. Sequence homology (98.5% (97.3%-99.3%)) was
observed within nested PCR. B) Nested qPCR retargeted to the region encompassing HDAg transcript and genome was used to
confirm the viral sequence present in a second pSS cohort and in a selection of patients experiencing sicca symptoms but did not
meet full criteria for diagnosis. Two samples identified as positive and two samples identified as negative by qPCR performed on
UK co-hort were independently processed and utilized as positive and negative controls in independent confirmation of HDV by
nested qPCR in additional UK co-hort.
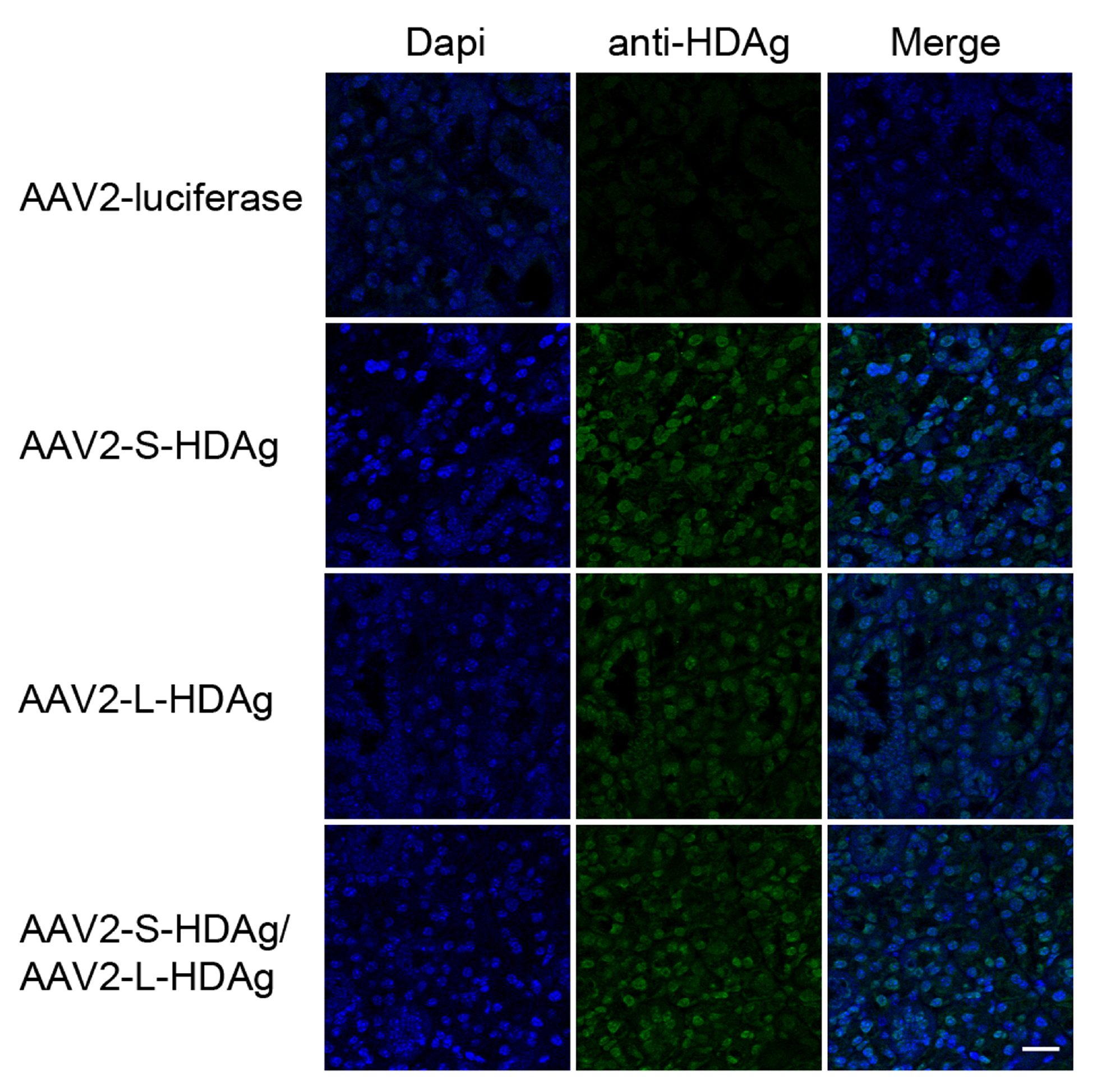
Supplemental Figure 2. Detection of HDAg in mouse salivary gland tissue in mice cannulated with AAV-HDAg. Retrograde cannulation was used to delivery recombinant adeno-associated virus serotype 2 (rAAV-2) containing HDV genes to salivary gland tissue in female C57BL/6 mice. Salivary gland tissue was evaluated 4 months post cannulation for expression of HDAg (green), and dapi (blue) for nuclear localization. Scale bar 20μm
<a href="https://www.paijournal.com/index.php/paijournal/article/download/72/version/17/36/742/72-289-1-SP-10.jpg" target="_blank">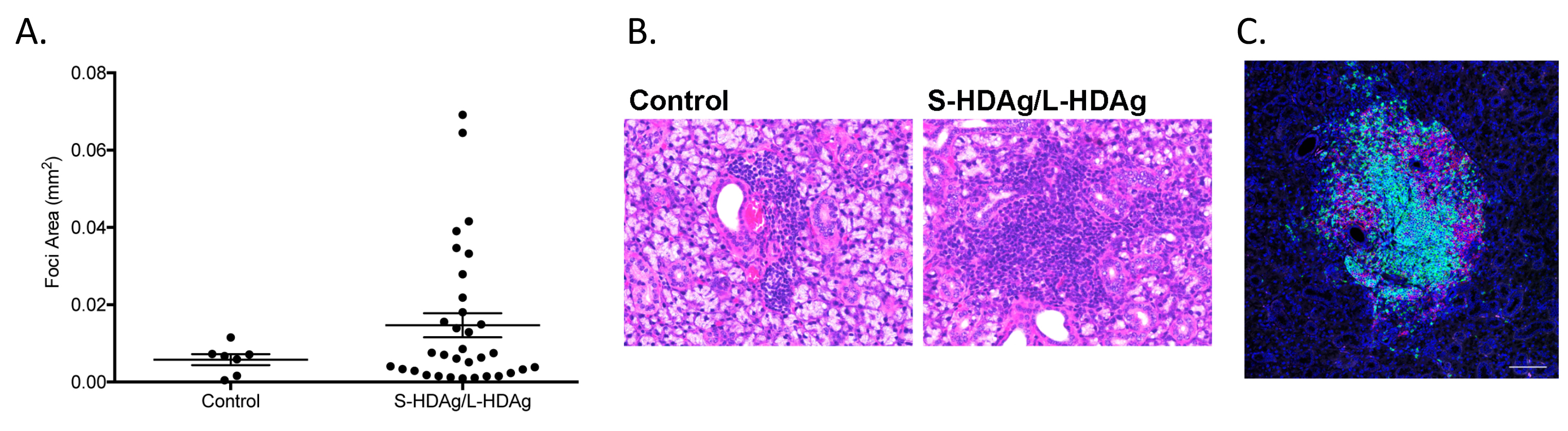
<
Supplemental Figure 3. Focal size of lymphocytic infiltrates were larger in mice expressing the combined S‐HDAg and L‐HDAg compared to foci size observed in mice expressing S‐HDAg/L‐HDAg in salivary gland tissue. A) Area of foci in mice expression a combination of both the small and large HDV antigen revealed an increase in size a subset of the observed foci (n.s., n = 7‐33). There was a significant increase in variance of size in the treated mice compared to the control mice (F‐test, P = 0.0008). B) Lymphocytic foci representative of the average size of foci present in control mice that expressed luciferase in salivary gland tissue (2.5E‐3 mm2). Average size of foci in the mice expressing S‐HDAg/L‐HDAg is 13.1E‐3mm2 in control mice. Images of lymphocytic foci representative of the average size of foci present. C) Ectopic lymphoid structures were evident in mice expressing combined S‐HDAg/L‐HDAg. Distribution of T‐cell CD3 (green) and B‐cell B220 (red) depict transition to organized lymphoid‐like structures (bar 100um).