Panagiotis Zagaliotis1,5, Jordyn Michalik-Provasek2, Jason J. Gill2, and Thomas J. Walsh1,3,4
1Transplantation-Oncology Infectious Diseases Program, Weill Cornell Medicine New York, NY
2Center for Phage Technology, Texas A&M University, College Station, Texas
3Departments of Pediatrics and Microbiology & Immunology, Weill Cornell Medicine New York, NY
4Center for Innovative Therapeutics and Diagnostics, Richmond, VA
5Department of Pharmacology and Therapeutics, School of Pharmacy, Aristotle University of Thessaloniki, Greece
Thomas J. Walsh, MD, PhD (Hon), FIDSA, FAAM, FECMM
tjwalsh@CITDx.org
Panagiotis Zagaliotis, MS
paz4002@med.cornell.edu
Zagaliotis P, Michalik-Provasek J, Gill JJ, Walsh TJ. Therapeutic Bacteriophages for Gram-Negative Bacterial Infections in Animals and Humans. Pathogens and Immunity. 2022;7(2):1–45. doi: 10.20411/pai.v7i2.516
10.20411/pai.v7i2.516
Drug-resistant Gram-negative bacterial pathogens are an increasingly serious health threat causing worldwide nosocomial infections with high morbidity and mortality. Of these, the most prevalent and severe are Pseudomonas aeruginosa, Klebsiella pneumoniae, Escherichia coli, Acinetobacter baumannii, and Salmonella typhimurium. The extended use of antibiotics has led to the emergence of multidrug resistance in these bacteria. Drug-inactivating enzymes produced by these bacteria, as well as other resistance mechanisms, render drugs ineffective and make treatment of such infections more difficult and complicated. This makes the development of novel antimicrobial agents an urgent necessity. Bacteriophages, which are bacteria-killing viruses first discovered in 1915, have been used as therapeutic antimicrobials in the past, but their use was abandoned due to the widespread availability of antibiotics in the 20th century. The emergence, however, of drug-resistant pathogens has re-affirmed the need for bacteriophages as therapeutic strategies. This review describes the use of bacteriophages as novel agents to combat this rapidly emerging public health crisis by comprehensively enumerating and discussing the innovative use of bacteriophages in both animal models and in patients infected by Gram-negative bacteria.
Keywords: bacteriophage; phage therapy; Gram-negative bacteria; antibacterial resistance; multidrug resistance
Following the discovery of penicillin, the world of medicine entered “the golden era of antibiotics” [1] . Infections could be treated and kept under control by antimicrobial agents with unprecedented efficacy. However, the very success of antibiotics, as well as their broad availability, led to their widespread use and to the subsequent development of antimicrobial resistance by many pathogens [2]. Among the additional causes of antibiotic resistance are their inappropriate prescribing, extensive use in agriculture, and relatively small number of new antimicrobial agents with novel mechanisms of action [3].
The World Health Organization (WHO) has established a list of high priority pathogens on which research and development of new antimicrobial agents are imperative [3]. The 5 critical-priority pathogens are the Gram-negative bacteria Acinetobacter baumannii, Pseudomonas aeruginosa, and carbapenem-resistant Enterobacteriaceae (eg, Klebsiella pneumoniae, Escherichia coli, and Enterobacter spp.), while Salmonella spp. are considered of high priority [4].
Gram-negative bacteria are particularly difficult to treat due to the presence of the outer cell membrane (containing lipopolysaccharide/endotoxin), which negatively affects permeability to antibiotics and is a major determinant of intrinsic antibiotic resistance [3]. Gram-negative rods are responsible for several types of infections, such as intraabdominal infections (IAIs), urinary tract infections (UTIs), and hospital-acquired pneumonia. More than 25% of all pathogen-related nosocomial infections in the United States are caused by these bacteria, with P. aeruginosa, K. pneumoniae, and E. coli infections constituting nearly 70% of all Gram-negative-related infections [5]. This renders Gram-negative infections an ongoing public health risk, which, in combination with antibiotic resistance, elevates the need for potential treatments for these infections.
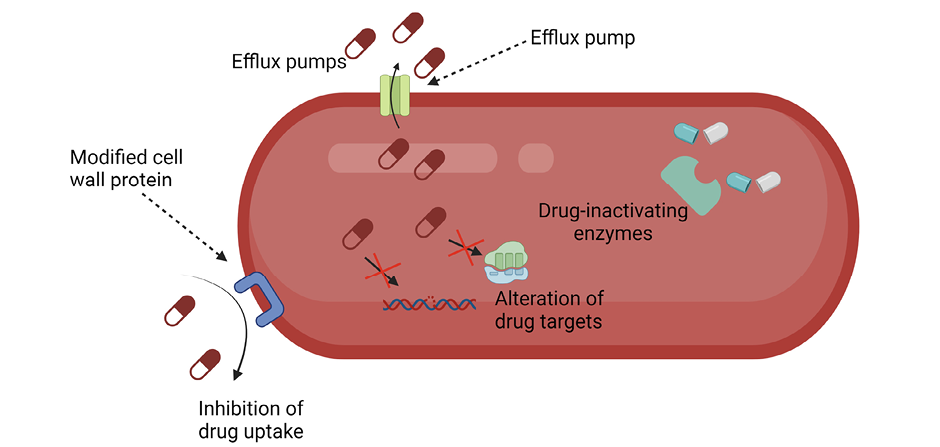
In addition to the intrinsic antibiotic resistance due to their cell envelope structure, Gram-negative bacteria possess several other mechanisms of antibiotic resistance (Figure 1A). Among these mechanisms are limiting the uptake of a drug, modification of a drug target, inactivation of a drug, and active efflux of a drug [6].
The clinical utility of new antimicrobial agents designed to treat resistant bacteria is being eroded. For example, cefiderocol with its novel catechol-based iron transport mechanism for delivering a beta-lactam antimicrobial agent to the cell wall of multidrug-resistant Gram-negative bacteria may be threatened by a wide range of mechanisms of resistance [7]. Among these mechanisms are novel beta-lactamases, efflux pumps, porin mutations, and mutations of siderophore receptors, as well as modified penicillin-binding proteins. Heteroresistance among strains of Acinetobacter baumannii poses additional challenges to antimicrobial therapy by permitting a subpopulation of resistant organisms to emerge during the course of treatment and to transmit genetic information to the susceptible population [7].
Pathogens can be classified as multidrug resistant (MDR), extensively drug resistant (XDR), and pandrug resistant (PDR). While definitions for these types of resistance categories have varied, an initiative by the European Centre for Disease Prevention and Control (ECDC) and the Centers for Disease Control and Prevention (CDC) concluded with the following definitions: MDR was defined as acquired non-susceptibility to at least 1 agent in 3 or more antimicrobial categories, XDR was defined as non-susceptibility to at least 1 agent in all but 2 or fewer antimicrobial categories (ie, bacterial isolates remain susceptible to only 1 or 2 categories), and PDR was defined as non-susceptibility to all agents in all antimicrobial categories [8]. The bacteria examined in this review are either MDR or XDR, with some cases of PDR A. baumannii, K. pneumoniae and P. aeruginosa having also been reported [9] .
Given the increasing resistance of bacteria to antibiotics and the relatively low number of newly available antibiotics, we have entered the “post-antibiotic era” [10] , where bacterial infections are very difficult or sometimes impossible to treat with existing antibiotics. Therefore, the need for new treatment options is urgent. One such option is the use of bacteriophages, which are viruses that infect bacteria.
Bacteriophages were first discovered in 1915 by Frederick Twort, an English physician [11]. Two years later, the French-Canadian microbiologist Felix d’ Herelle independently published similar observations, and would continue working with bacteriophages for most of his career [12]. Bacteriophages act by causing lysis of the bacterial cell with the recruitment of several mechanisms [13]. These mechanisms dictate the life cycle of the phages, which can be either lytic or lysogenic (or temperate)(Figure 1B). The mechanisms entail the use of holins to create pores in the cell membrane, as well as endolysins to degrade the peptidoglycan of the cell wall of Gram-negative and Gram-positive bacteria. These 2 types of enzymes are often sufficient to disrupt the bacterial cell envelope (cell membrane and cell wall) of Gram-positive bacteria. A third class of bacteriophage enzymes, spanins, is needed for lysis of Gram-negative bacteria. These enzymes fuse the inner and the outer cell membranes of Gram-negative bacteria, therefore causing lytic disruption [13].
In most cases, bacteriophages are administered to animals in the form of whole phage particles. Certain studies have also reported the use of bioengineered phage products and individual phage enzymes, such as endolysins, which are peptidoglycan-degrading enzymes that normally take part in the lysis of the bacterial cell wall [14–17].
Bacteriophages are relatively easy to isolate from the environment, with several methods having been reported [18]. Furthermore, they exhibit high host specificity, usually targeting only specific strains of bacteria and leaving the microbiota as well as eukaryotic cells unaffected. In addition, they have the advantage of self-replicating at the expense of their hosts, in contrast to conventional small molecule antibiotics.
Figure 1A. Mechanisms of bacterial resistance to antibiotics.
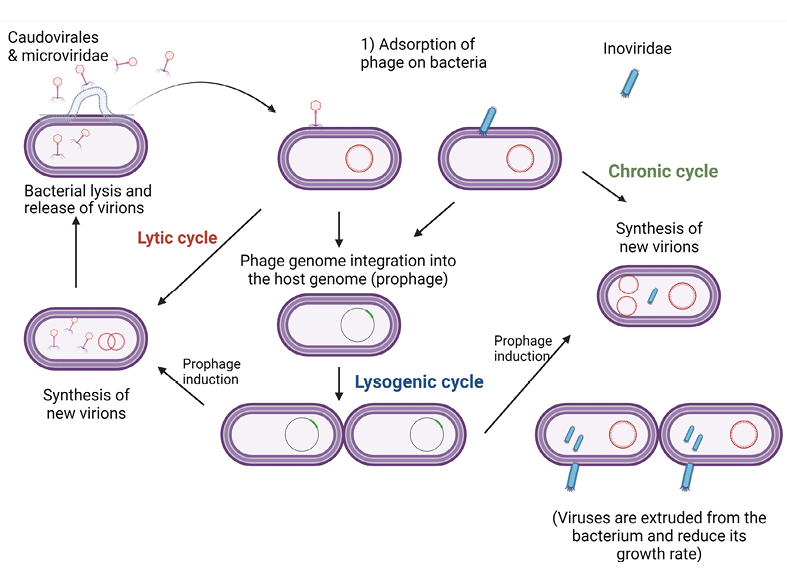
Figure 1B. Life cycles of phages. Bacteriophages can either be lytic, in which case they cause lysis of the bacterial cell and release progeny within a short period of time, or lysogenic, in which case they inject their genome into the bacterial genome and stay dormant for a while until they get activated and release progeny. For therapeutic purposes, the preferred type of phage is lytic, as killing of the bacteria within a short period of time is necessary in treatment of bacterial infections.
As with many novel antibacterial treatment efforts however, the fact that many bacteriophages have shown in vitro activity does not automatically ensure their in vivo efficacy. Factors such as systemic clearance, pH, and route of administration increase the unpredictability of otherwise successful in vitro applications. Therefore, the objective of this article is to review the in vivo and clinical applications of phage therapy against Gram-negative bacterial infections, alone or in combination with antibiotics. Even though several review articles have been written with regard to phage therapy, they either discuss phage therapy as part of potential treatment alternatives against the so-called ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp) [19], or they do not discuss extensively the aspects of phage kinetics, potential immune reactions, and histology outcomes of phage therapy [20]. This review further discusses the advantages, potential limitations, and potential developments that will optimize the future applications of phage therapy.
The use of bacteriophages has been reported in many studies of different animal models, mimicking the infections that can occur in humans. Translatable models of mice and rat infections have been used more widely [15, 16, 21–46], with infection models in rabbits, swine, sheep, or cattle having been used less frequently [47–51].
Most studies focus on the therapeutic efficacy of phage, but many also address phage kinetics, ie, the rate of clearance from the bloodstream and tissues [21–23, 26, 29, 52–55]. As phages have the ability to self-replicate and can be cleared at different rates according to conditions found in the tissues, these important pharmacokinetic aspects of phage therapy differ greatly from those of conventional antimicrobial agents. The route of administration also impacts the pharmacokinetics of bacteriophage in vivo. Gastric pH or the need to cross the gastrointestinal epithelium may limit the utility of oral administration of bacteriophage for systemic therapy. Other studies also examine the effect of phage on the infected tissues, as well as on the immune response of the host, by measuring pro-inflammatory cytokines, such as TNF-α and IL-6 [21–23, 26–28, 40, 42, 45, 46, 53, 55–59]. Measurements of these pro-inflammatory cytokines may serve as surrogate biomarkers for Gram-negative sepsis. The temporal patterns of serum pro-inflammatory cytokines generally parallel improvement in survival and reduction of residual bacterial tissue burden in animal models.
In studies of potential synergy between phage and host immune response, both immunocompetent and immunosuppressed animals were used to compare the differences in outcome [29].
Gram-negative bacilli are important causes of hospital-acquired pneumonia and chronic lung infections worldwide, with many animal models for treatment of Gram-negative bacillary pneumonia having been developed [21, 22]. Table 1 summarizes the in vivo studies of therapeutic bacteriophages against Gram-negative bacterial pneumonias.
In most cases, intranasal inoculation is the preferred route of administration [13–27, 52, 53, 56, 60, 61]. This route ensures that the phage would reach the bacteria in the lungs in high concentrations and achieve a faster therapeutic outcome. Some models also use the inhalational or intraperitoneal routes [28–30, 47, 54, 55, 62]. Independent of the route of administration, most phage doses range from 107 plaque-forming units (PFU) to 1010 PFU, and all models include at least 1 dose that is 10-fold higher than that of the bacterial burden at the time of administration. This ratio allows for a multiplicity of infection (MOI) of 10. Singla et al [47] reported the only study that used a lower dose (104 PFU), as it aimed to examine the effect of liposomal entrapment of the phage on the therapeutic outcome. The phage dose for the liposome-entrapped phage in that study was as low as 102 PFU.
The outcome of treatment in these studies was determined by both the dose and route of administration, as well as the timing of the dosing. Both survival and reduction of colony-forming units (CFU) were dose-dependent and were improved at MOIs ≥ 10, while a better outcome was achieved for quicker initiation in therapeutic models.
All studies that examined bacteriophage kinetics showed that phage titers were higher at the same timepoints in bacteria-infected groups than in phage-only groups (animals free from bacterial infection), both in tissue and in blood. This indicates that phage population expands when encountering and replicating in host bacteria, leading to higher in vivo concentrations in infected animals.
Histopathological examination of the tissues in some studies revealed that phage treatment may also protect lungs of treated animals from alveolar wall thickening and neutrophil infiltration, compared to untreated controls [23, 53–56, 60].
Table 1. Animal Pneumonia Studies
|
Reference |
Pathogen |
Animal Model |
Dose & Route of Administration |
Outcome |
Phage Kinetics |
Histology & |
|
[56] |
A. baumannii |
Mouse pneumonia |
Single phage dose, IN |
>2 log10 reduction in CFU by day 1, bacteria were cleared from lungs by day 3. Treated groups survived as follows: a) MOI = 10, 100%, |
Treatment group (lungs): Phage-only group: |
Untreated group: Severe alveolar wall thickening, hemorrhaging in alveolar space. Treated group: Only mild to moderate thickening of the alveolar walls. |
|
[23] |
A. baumannii |
Mouse pneumonia |
3x105 / 3x106 / 3x107 PFU/mouse, IN |
3 log10 reduction for treated group, 24 hpi. P<0.0001 MOI = 10 increased survival by 90%. |
Phage-only group (no bacteria): 4.22x106 PFU/lung Phage-treated group (bacterial infection): 8.3x107 PFU/lung. 1log difference at 24 hpi. |
Non-treated group: severe thickening of alveolar wall, severe cell infiltration of perivascular and peribronchial region. Phage-treated group: no inflammation, milder thickening of alveolar wall |
|
[52] |
A. baumannii |
Mouse pneumonia |
108 PFU/mouse IN, Q=24h |
2 log10 reduction in CFU between treated and non-treated group on days 3 and 4. Treated group – 35% survival, non-treated group – 15% survival 7 dpi |
N/A |
Immune responses to phage-only: 20% increase in serum IgE, slight increases in serum GM-CSF, IL-2, IL-10, and IL-17α, after IP, IN, and oral administration |
|
[53] |
A. baumannii |
Mouse pneumonia |
2x108 PFU/mouse, IN |
4 log10 reduction in CFU in the treated group, compared with the non-treated one, 3 dpi. Treated group: 100% survival at 6 dpi. Untreated group: 0% survival at 6 dpi. |
Phage-only group: 2 log10 less PFU than the infected group in each time point (ie,12h, 24h, etc.) |
Non-treated mice: heavier rates of congestion and edema than phage-treated mice |
|
[24] |
E. coli |
Mouse pneumonia |
|
3 log10 reduction in CFU/g with both routes |
Phage counts were the same in 2 groups 17h post infection, even though IP dose was initially 10 times higher |
N/A |
|
[60] |
E. coli |
Mouse pneumonia |
MOIs 0.3, 3, or 10 (1.2x107, 1.2x108 or 4x109 PFU/mouse), IN |
100% survival in treated mice on day 3, compared with 0% in control group on day 3, at all MOIs. Adaptation of the phage to the VABP strain increased survival from 20% to 75% |
1.1 log10 increase in PFU counts from the 6h to the 16h mark after infection |
PMN engulfment in BAL fluid and chemokine significantly lower in phage-treated group, 16h post infection |
|
[25] |
P. aeruginosa |
Mouse pneumonia |
900μg/mouse, IN or IT |
20% survival for IN administration, 70% survival for IT administration (10-day period) |
N/A |
N/A |
|
[61] |
P. aeruginosa |
Mouse pneumonia |
106 or 2x107 PFU/mouse, IN |
100% survival with PhiKZ phage at MOI = 20, 0% at MOI = 0.1 90% survival with PAK_P1 at MOI = 0.1 |
N/A |
N/A |
|
[26] |
P. aeruginosa |
Mouse pneumonia |
2-5x109 PFU/mouse, IN |
3 log10 decrease in CFU in treated mice |
N/A |
N/A |
|
[27] |
P. aeruginosa |
Mouse pneumonia |
2x107 PFU/mouse, IN, 2 doses at 24 and 36h post infection OR at 48 and 60h post infection OR at 6 days post infection |
3 log10 reduction in CFU/g in lungs of treated mice, with complete bacterial clearance eventually, in the first 2 groups Complete clearance in 70% of mice and 1 log10 reduction in the remaining 30% in treated mice 6 days post infection, in third group |
N/A |
N/A |
|
[47] |
K. pneumoniae |
Mouse pneumonia |
Therapeutic use: Liposome entrapped phage: 102 PFU/mouse, IP, 6h, 24h, 48h, and 72h post infection Prophylactic use: Liposome entrapped phage: 102 PFU/mouse, IP, 6h, 24h, 48h, and 72h prior to infection |
Therapeutic use: Liposome entrapped phage group: 6h and 24h doses cleared all bacteria. Prophylactic use: Liposome entrapped phage group: 6h, 24h, and 48h doses before infection, completely inhibited infection. 72h dose did not have any effect |
Non-infected mice which were given phage had higher retention than infected ones for up to 3–4 days |
Phage-treated group: IL-1β and TNF-α reduced in phage- treated group, either 6h prior to infection or after infection. IL-10 was increased in treated group. Liposome-entrapped phage group: Cytokine levels displayed more significant difference compared with both control group and phage-only group. Doses at all times lead to the differences, except for dose 72h prior to infection. Both prophylactic and therapeutic dose protected lungs from neutrophil infiltration and damage. |
|
[54] |
K. pneumoniae |
Mouse pneumonia |
109 ( MOI=10) or 1010 (MOI=100) PFU/mouse, IP |
MOI = 10 resulted in 5 log10 reduction in CFU 3 days post infection ΜΟΙ = 100 resulted in 7 log10 reduction |
N/A |
No lung enlargement or abscesses in treated groups |
|
[28] |
P. aeruginosa |
Mink pneumonia |
108, 109, and 1010 PFU/mink, by ultrasonic nebulization |
1010 PFU reduced CFU by 1.5 log10 and 80% survival |
N/A |
N/A |
|
[29] |
P. aeruginosa |
Mouse pneumonia |
108 PFU/mouse, inh, at 2h after infection |
100% survival in treated mice compared with 0% in untreated ones, as well as complete clearance of bacteria 48h after treatment. Intact innate immunity was required for phage treatment success. Neutropenic mice could not be saved even with 100-fold less bacterial inoculum |
Phage was cleared from mice at a rate of 0.5 log10/day, which resulted in efficacy of treatment up to 4 days after phage administration (MOI = 0.1 at that point) |
N/A |
|
[55] |
P. aeruginosa |
Mouse pneumonia |
107 PFU/mouse (MOI = 10), IP |
Phage-treated groups: 66% and 83% survival on day 12, compared with 0% survival (all died by day 3) in untreated group 1–2 log10 reduction in CFU/g in lungs with both phages on day 1, 6 log10 reduction from both phages on day 5, with some mice undergoing complete clearance of bacteria on day 5 |
N/A |
Untreated group: severe alveolar wall thickening and neutrophil infiltration. Treated group: Mild or no histological findings. TNF-a and IL-6 levels equal on day 1, but significantly reduced on day 5 in treated group |
|
[62] |
P. aeruginosa |
Mouse pneumonia |
107 PFU/mouse, inh, single dose at 2h post infection |
5 log10 reduction in CFU/g in lungs at 24h |
Phage steadily increased in the first 24 hours, reaching 1–2 log10 higher concentrations at 24h after administration in the lungs and plasma |
N/A |
|
[30] |
E. coli |
Chicken pneumonia |
Approx. 109 PFU/mL, aerosolized, or IM, 2h, 24h, or 48h after bacterial inoculation |
Aerosol administration directly after E. coli infection increased survival from 50% to 80%. |
Aerosol: a few birds had an average of 102 PFU/mL1h after administration, no phage at 24h and 48h. |
Non-treated birds: colibacillosis lesions, pericarditis and increased spleen and liver weights |
Abbreviations: CFU: colony forming unit; PFU: plaque forming unit; MOI: multiplicity of infection; IN: intranasal; IT: intratracheal; dpi: days post-inoculation; N/A: not applicable
The presence of bacteria in the bloodstream (bacteremia) can cause sepsis and septic shock, especially when a patient is immunocompromised [31]. Animal models of bacteremia have used phages to treat this potentially fatal condition (Table 2).
The most common route of administration in such studies is intraperitoneal or intravenous injection [16, 24, 32–37, 63–65]. Four studies also examined the use of isolated phage peptides, rather than whole phage particles, for the treatment of bacteremia in mice and rats [17, 19, 38, 39]. In all these studies, intraperitoneal injection is the preferred route of administration. All phage doses aimed for MOIs ≥ 1, while Wang et al [65] explored the efficacy of phage treatment at MOIs ranging from 0.0001 to 200.
|
Reference |
Pathogen |
Animal Model |
Dose & Route of Administration |
Outcome |
Phage Kinetics |
Histology & |
|
[14] |
A. baumannii |
Mouse bacteremia |
108 PFU/ml, IP |
Treated group: 100% survival at 6 weeks Untreated group: 0% survival |
N/A |
N/A |
|
[15] |
K. pneumoniae |
Mouse bacteremia |
Prophylactic treatment: Depolymerase 50μg/mouse, IP, single dose 6h prior to infection. Therapeutic treatment: Depolymerase 50μg/mouse, IP, single dose 30 min after infection |
100% survival in mice treated with Dp42 in both prophylactic and therapeutic treatment. |
N/A |
N/A |
|
[17] |
E. coli |
Rat bacteremia |
0.25 μg of depolymerase, IP single dose |
Untreated group: 0-20% survival. |
N/A |
N/A |
|
[24] |
E. coli |
Mouse bacteremia |
6x1010 PFU/mouse IP, single or double dose |
Single or double dose was not able to prevent the death of animals, but resulted in a ~1.5 log10 reduction in CFU/gr |
Phage counts were 1 log10 higher in double dose group than in single dose group |
N/A |
|
[30] |
K. pneumoniae |
Mouse bacteremia |
1.75x108 PFU/mouse, IP, single dose |
Untreated group: 0% survival. Phage-treated group: 100% survival when phage was administered 10 min after bacterial challenge. |
N/A |
N/A |
|
[32] |
A. baumannii |
Mouse bacteremia |
5x108 PFU, IP, Q=24h for 6 days |
Phage-treated group: 100% survival. Untreated group: 10% survival. No bacteria in kidneys and liver in treated group. High bacterial burden (109 CFU/gr) in untreated group |
N/A |
N/A |
|
[65] |
E. coli |
Mouse bacteremia |
From 103 to 6x109 PFU/mouse, IP, single dose |
With a MOI from 10-4 to 200, 100% survival rate. Lower MOIs (10-5 and lower) decreased survival to 0-20% |
109 PFU/mL were retained up to 24h after injection, 106 PFU/mL 48h after |
N/A |
|
[33] |
E. coli |
Mouse bacteremia |
108 PFU/mouse, IV, single dose at 10 min, 1h, or 2h after infection |
95-100% survival if administered within 1h, 33% survival if administered within 2 hours. No untreated mice survived. |
Phage was detected up to 2 weeks after infection and administration. |
N/A |
|
[35] |
E. coli, K. pneumoniae and P. aeruginosa |
Mouse bacteremia |
109 PFU/mouse, IP at 5h, 14h, and 18h after infection |
100% survival in all pathogen-infected mice after 5 days, compared with 0% in control. |
N/A |
N/A |
|
[64] |
P. aeruginosa |
Mouse bacteremia |
5x106, 5x107, or 5x107 PFU/mouse, IP, single dose |
Immunocompetent mice: 100% survival with MOIs of 10 and 100 (5x107 and 5x108 PFU); 80% survival with MOI = 1. |
Phage was cleared from all organs in both infected and non-infected mice within 72h. In infected mice, phage persisted in the spleen for up to 96h |
N/A |
|
[36] |
P. aeruginosa |
Mouse bacteremia |
109 PFU/mouse, IP, single dose |
Phage-treated group: 92% survival Untreated group: 7.4% survival. |
Phage titers increased in blood, liver, and spleen during the first 12h and then decreased. Cleared from blood within 24h and from both blood and liver at 48h |
N/A |
|
[63] |
P. aeruginosa |
Mouse peritonitis - sepsis |
2x108 PFU/mouse, IM or IP, single dose at 6h post infection (2 different phages were administered) |
Survival with both phages was 100% or 80%, compared with 0% in untreated group |
Phage were detectable in blood, liver, and lung up to 36h post infection. They were cleared faster from blood and lung compared with liver. |
N/A |
|
[37] |
P. aeruginosa |
Mouse bacteremia |
104 PFU/mouse, IV, single dose at 1h after infection |
8 log10 reduction in CFU after 2.5h, compared with non-treated mice. Lower concentrations than 104 were ineffective in treatment of the infection. |
Phages remained in the liver and spleen for 36h in healthy mice, but were minimally detectable in urine. |
N/A |
|
[38] |
P. aeruginosa |
Mouse bacteremia |
Lysocin 2.5, 5, 12.5, and 25 mg/kg, IP, 3h post infection |
73%, 80%, 93%, and 100% survival with each dose respectively, compared with 37% survival of control group |
N/A |
N/A |
|
[39] |
A. baumannii |
Mouse bacteremia |
1mg of lysin, transcatheter, 2 doses, 4 hours apart |
2log10 reduction in catheter-mimicking model, with phage administration 24 hpi Phage-treated group: 50% survival. Untreated group: 10% survival |
Ν/Α |
Ν/Α |
Abbreviations: IP: intraperitoneal; PFU: plaque forming unit; N/A: not applicable; IM: intramuscular; IV: intravenous; CFU: colony forming unit
As with pneumonia, the result of phage treatment was both dose-dependent and time-dependent, with the time of initiation of the treatment being more important in rescuing the animals. Higher survival rate was inversely related to time of initiation of phage therapy. This effect was observed with either the use of peptides (such as lysins) or whole phage particles in cocktails. Notably, no dose of bacteriophage was sufficient to rescue animals if treatment started too late, as shown by Horvath et al [34], while similar observations were made in the case of depolymerase (lysin) administration [19]. The immune system of the animals is another important aspect which plays a role in treatment outcome. As previously stated [64], immunocompetent mice achieved high survival rates compared with neutropenic mice, none of which survived in the study of bacteremia caused by P. aeruginosa. As with the other studies, the result of treatment was also dose-dependent, with MOIs of 10 and 100 showing 100% survival, compared with 80% survival with MOI of 1. The results of phage therapy in different laboratory animal models are a function of inoculum size, level of immunosuppression, genetics of the animal, timing of initiation of therapy, and dosage schedule of the intervention. When such conditions are optimized, phage therapy and antibacterial therapy may be highly effective in treating the target infectious disease.
For kinetics, the existence of bacteria in the bloodstream was important for the retention of phage, with non-infected animals clearing phage faster. Phage also persisted longer in the spleen and secondarily in the liver, compared with other organs or the blood [33, 36, 37].
Infections of open wounds by Gram-negative bacilli present an urgent necessity for treatment, as they can cause locally progressive tissue injury and bacteremia. Burns, trauma, and pressure injuries are especially challenging infections in hospitalized patients. Most animal models that address these infections in the preclinical setting involve the use of mice [37–39, 55, 63, 66, 67], with one study using chickens as well as mice [41] (Table 3).
Wound dressings are used to apply bacteriophages for topical administration in these infections. Two models, one of which examined the effect of liposome encapsulation on the efficacy of the phage, used intraperitoneal injections [37, 67]. All doses fell within 5x106 – 1010 PFU for whole phage particles. Chadha et al [57], who examined the effect of liposome encapsulation of the phage, used 105 PFU for both phage-only and liposome-entrapped phage cocktails, while Raz et al [25], who used a phage lysin, administered 100-300 μg/mouse topically.
Table 3. Animal Wound Infection Studies
|
Reference |
Pathogen |
Animal Model |
Dose & Route of Administration |
Outcome |
Phage Kinetics |
Histology & |
|
[25] |
P. aeruginosa |
Mouse wound infection |
Lysin, 100, 200, or 300 μg/mouse, topically, single dose |
100 μg – 1 log10 reduction in CFU 300μg – >2 log10 reduction in CFU |
N/A |
N/A |
|
[66] |
A. baumannii |
Mouse wound infection |
5x108 PFU, SC, or by direct pipetting on the wound |
Wounds in local application of phage were significantly smaller than non-treated group and systemic application (differences between systemic-application group and non-treated group were minimal) |
N/A |
N/A |
|
[67] |
A. baumannii |
Mouse wound infection |
108 PFU/mouse, IP, and 5x107 PFU/mouse topically in Tegaderm dressing, at 4h, 24h, and 48h post infection |
Non-treated mice did not see reduction or closure of wound. The 3rd group, non-prophylactic treatment and phage post infection, showed the highest reduction in wound size and the wound had closed by day 17 |
24h following 108 PFU/mouse IP administration, the spleen had 105 PFU/g, the liver had 103 PFU/g and the lymph nodes had from 103 - 5x107 PFU/gr. Control group had no detectable phage in any of these organs |
N/A |
|
[40] |
E. coli |
Mouse wound infection |
109 PFU, topically |
No difference in CFU counts was observed in MG1655 strain. |
N/A |
N/A |
|
[58] |
E. coli |
Mouse wound infection |
1010 PFU/mouse, single dose, topically |
A single dose of phage had the same efficacy as repeated doses of antibiotics and had cleared bacteria from the wound by day 3 |
N/A |
N/A |
|
[41] |
K. pneumoniae |
Mouse wound infection |
5x106 PFU/mouse, topically. 7 groups of mice: One control, 5 with single phage and one with a cocktail of phages |
Untreated group CFU/g: 108.8 |
Similar pattern for all groups: |
Untreated group: complete destruction of superficial skin layers along with partial fusion of collagen and dense inflammation. Significant increase in neutrophil infiltration and profound inflammation was observed in the skin. Phage-treated groups: healing in the form of collagen and fibroblastic proliferation and regeneration of epidermis with mild infiltration of lymphocytes. |
|
[57] |
K. pneumoniae |
Mouse wound infection |
Group 1: Untreated control Group 2: Phage 105 PFU, IP Group 3: Liposome entrapped phage 105 PFU, IP Group 4: Liposome alone, IP, all single dose |
Bacterial counts kept increasing in untreated and liposome-only (control) groups for up to 72h, in blood, skin, and liver. Phage-treated and liposome-phage groups showed continuous reduction at 24h, 48h, and 72h, between 4 and 6 log10 reduction, with liposome entrapped phage group showing about 1 log10 lower counts than phage-only group at all timepoints |
Phage alone remained in liver, spleen, and blood up to 36h after injection(peak at 6h) Liposome entrapped phage persisted in these organs up to 72h (peak at 6h) and up to 120h in the spleen (102.5 PFU/gr) |
Untreated animals (control) showed loss of epidermis and massive infiltration of neutrophils extending up to the muscle layer. Loss of distinction among individual collagen fibers. Thermally injured and infected mice, receiving CP showed regeneration of epidermis along with mild infiltration of neutrophils. Maximum healing was observed in mice administered with LCP |
|
[68] |
K. pneumoniae |
Mouse wound infection |
5x109 PFU/mouse (MOI = 200), topically, single dose |
63.33% survival in treated group at 7 days compared with 0% in control group |
N/A |
|
|
[69] |
P. aeruginosa |
Mouse wound infection |
Topically via nanofibrous dressing, single dose (dose not mentioned) |
Wound size started decreasing on day 1 with all agents used, including antibiotics and phages, and the wound was completely healed on day 12 from either one |
N/A |
Increased collagen deposition and faster dermis formation on the site of the wound in the phage-treated group, compared with the untreated one |
Abbreviations: IP: intraperitoneal; PFU: plaque forming unit; N/A: not applicable; IM: intramuscular; IV: intravenous; CFU: colony forming unit; SC: subcutaneous
In all models, a complete healing or significant reduction in the size of the wound was observed, with bacterial colony forming units (CFU) on the wound reduced 10-fold to 105-fold. Liposome entrapment also increased the efficacy of treatment, leading to a greater reduction in CFU compared with the phage-only group [67]. The use of a phage cocktail composed of 2 or more bacteriophages was also shown to have a better impact on the outcome of the treatment when compared with single phage administration [66].
Three of the studies included an analysis of phage kinetics, 1 for topical administration [66] and the other 2 for intraperitoneal (IP) injection [37, 67]. In IP administration, the spleen was the organ with longest phage persistence, followed by the liver, while the entrapment of phages in liposomes reduced their clearance, maintaining them longer in the spleen, liver, and other tissues.
In those studies that histologically examined wounds [41, 58, 67], phage administration reduced the extent of inflammation and superficial skin destruction, as well as neutrophil infiltration, while liposome entrapment enhanced this activity.
Gram-negative bacilli are responsible for other infections, such as those involving the gastrointestinal (GI) tract, brain, cornea, the urinary tract, and muscle. Models of bacteriophage therapy for these conditions have been investigated and are reported in Table 4.
The route of administration in these studies depends on the type of infection; eg, orally or intraperitoneally administered bacteriophages are used to treat GI tract infections and urinary tract infections (UTIs) [42, 43, 60, 63]. Eye drops are used for bacterial keratitis [69] and intramuscular administration is used for soft tissue infections [70–72]. Phage doses range from 107 – 1011 PFU/animal. Notably, a study by Bull et al [70] administered a dose as low as 100 PFU/animal intramuscularly in an E. coli thigh infection model, to examine the effect of the K1 antigen-specificity of the phage in the therapeutic outcome, showing that phages requiring the K1 capsule (K1-dependent phages) rescue mice even when administered at doses 106-fold lower than the K1-independent ones.
The result of most treatments was either the increase in survival of treated animals, or the decrease in CFU compared with the non-treated controls. The exception to this observation is the study by Reynaud et al [73], studying a rabbit ileal loop model of E. coli infection, in which phage administration only managed to postpone the death of the animals for 3 days, but did not significantly increase the survival rate compared with the untreated group. Moreover, Jamalludeen et al [74] demonstrated a reduced duration and severity of E. coli diarrhea and lower CFU counts in a porcine model, compared with the untreated group.
Table 4. Other Animal Infection Studies
|
Reference |
Pathogen |
Animal Model |
Dose & Route of Administration |
Outcome |
Phage |
Histology & |
|
[16] |
E. coli |
Mouse thigh infection |
Three different types of depolymerase, 0.1, 0.4, or 1 mg/kg, IM |
10% survival for non-treated groups. |
N/A |
Clinical evaluation showed no signs of health decline, weight loss etc |
|
[24] |
E. coli |
Mouse UTI |
1010 PFU/mouse, IP |
2 log10 reduction in CFU/g in kidneys |
109 PFU/g in the kidneys, 48h post infection |
N/A |
|
[63] |
P. aeruginosa |
Mouse peritonitis |
2x108 PFU/mouse, ΙΜ or ΙP, single dose at 6h post infection (2 different phages were administered) |
Survival with both phages was 100% or 80% with D3112 phage compared with 0% in untreated group or with MP22 phage. |
Phage were detectable in blood, liver, and lung up to 36h post infection. They were cleared faster from blood and lung compared with liver. |
N/A |
|
[42] |
E. coli |
Mouse UTI |
3x1011 PFU/mouse (MOI = 60), IP |
90% survival of phage-treated group compared with 0% of control, 3 dpi |
Phage particles were detected in liver, kidney, spleen, and lungs 4h post infection, and remained relatively stable up to 24h post infection in these organs. |
N/A |
|
[43] |
E. coli |
Rat GI infection |
7.2x107 PFU/rat, TD or orally |
0% survival in untreated group - 83.3% survival in treated group |
Infection-free rats: Transdermal administration: 1h after admin, phage titer in serum was high and remained elevated 24h after injection. |
Phage-treated group showed no damage or lesions. Untreated group: dense lymphomononuclear infiltration with oedema in lamina propria and inflammation of Peyer’s patch. No major alterations in kidneys, liver, and stomach. IL-6 was significantly reduced in phage-treated group |
|
[75] |
P. aeruginosa |
Mouse keratitis |
Topically (eye drops), single dose |
Mild or no corneal opacities in treated mice, in comparison with severe corneal perforation in untreated ones |
N/A |
Untreated mice: Destroyed stromal structure of cornea, severe neutrophil infiltration, high numbers of P. aeruginosa in the abscesses. |
|
[70] |
E. coli |
Mouse thigh infection |
K1 dep (dependent): 100 PFU/mouse, IM |
K1 dep saved 94% of mice at 100 PFU. |
K1-ind phages: 3 hpi: 108 PFU |
N/A |
|
[45] |
E. coli |
Mouse intramuscular and intracerebral infection |
of 3x108 PFU/mouse, IM or IV |
Intramuscular infection survival: non-treated group 7%, Intracerebral infection Survival: non-treated group 5%. |
Intramuscular infection |
N/A |
|
[73] |
E. coli |
Rabbit GI colonization |
109 PFU/rabbit via stomach tube |
Non-treated rabbits started dying on day 4. At the end of the experiment, 90% of animals had died. |
After administration of phage only, all the organs had 103-105 PFU/g phage until day 4. After day 7, phage in all organs was <10 PFU/g, except for the spleen, in which it remained around 103 PFU/gr. |
N/A |
|
[74] |
E. coli |
Pig diarrhea |
5 trials: Trial 2: 1010 PFU/pig, orally, but pigs were treated with antacids prior to challenge Trial 3: Prophylactic treatment, 15 min after bacterial inoculation |
Pigs that received phage mixture showed reduced duration of diarrhea and score of diarrhea (severity) in all trials. |
N/A |
N/A |
|
[76] |
E. coli |
Mouse peritonitis |
1011 PFU/mouse (one group with LyD - lysis deficient phage, the other with WT phage), IP |
81% survival in LyD-treated group, 55% in WT-treated, 33% in latamoxef-treated, no mice survived in control group 7 log10 reduction in CFU/g in all treated groups in 6 hours, 8 log10 reduction in 12 hours |
N/A |
TNF-α and IL-6 levels were not different at 6h, but LyD group had significantly lower TNF-α levels than all groups at 12h. IL-6 levels at 12h were lower than control group for all treated groups |
|
[77] |
K. pneumoniae |
Mouse liver abscesses |
2x105, 2x106, 2x107 or 2x108 PFU/mouse, oral or IP |
Untreated group: 10% survival CFU/g was not detected in liver or blood of treated mice at 6h, 24h, and 72h post infection |
N/A |
AST and ALT levels were lower in treated groups compared with untreated groups. TNF-α, IFN-γ, MCP-1, IL-10, and IL-6 were all at the same level as naive mice, compared with untreated group which had very elevated cytokine levels |
|
[78] |
E. coli |
Sheep GI infection |
1011 PFU/sheep, orally, single dose |
2 log10 reduction of CFU in caecum and rectum, 2 days after start of treatment |
N/A |
N/A |
|
[79] |
E. coli |
Rabbit ileal loop |
106 PFU/rabbit, orally |
Fluid accumulation in ileal loop was significantly inhibited in phage-treated group compared with untreated control. A 7 log10 reduction in CFU/g was also observed |
N/A |
N/A |
|
[80] |
E. coli |
Rat GI colonization |
Phage, 107 PFU/rat, in drinking water. Rats were separated in 2 trials: One only included the naturally inhabiting E.coli strains of rats, the other included inoculation with 60 human pathogenic strains |
1st trial (rat E. coli): For oral gavage phage: 1.8 log10 reduction, starting on day 2 to day 12. On day 12, CFU started to rise again and reached control group on day 20 Vegetable capsules: 3 log10 reduction on day 6. Started to rise again on day 12 and remained 1 log10 lower on day 20 2nd trial (human pathogenic E. coli strains): Oral gavage: Maximum reduction was on day 7, at 3.45 log10 Vegetable capsules: Maximum reduction 4.62 log10 on day 6. Then gradual increase back to baseline on day 20 |
Phage shedding peaked around 105 PFU between days 4-12 for all administration routes. Then it gradually declined until day 20 (0 PFU), following a reverse course than E. coli CFU counts. |
N/A |
|
[81] |
E. coli |
Mouse GI tract infection |
3 dosing regimens: Single administration of 108 PFU/mouse. |
Only daily administration of phage reduced bacterial burden in GI tract (1-2 log10) |
Daily administration pf phage kept phage concentration at 105 PFU/ml until day 5. |
N/A |
Abbreviations: IP: intraperitoneal; PFU: plaque forming unit; N/A: not applicable; IM: intramuscular; IV: intravenous; CFU: colony forming unit; GI: gastrointestinal; UTI: urinary tract infection.
With regards to phage kinetics, phages which encountered bacteria remained in tissues longer than those not encountering bacteria. Notably, no difference in phage clearance was observed in the study by Bull et al [70] between K1-dependent and K1-independent phage.
Three of the studies examined the histology or cytokine levels of animals in treated and untreated groups. In an E. coli peritonitis mouse model by Matsuda et al [76], IL-6 was significantly reduced in the phage-treated groups, whereas the use of a Lysis-Deficient (LyD) phage led to lower levels of TNF-α compared with mice with peritoneal infection treated with the wild type (WT) phage for E. coli. This was achieved due to the use of a phage mutant, which does not lyse bacterial cells, and therefore does not result in the release of endotoxin and consequent elevation of pro-inflammatory cytokine levels. It does, however, kill the bacteria, by cleaving their DNA with phage-induced enzymes, as well as using bacterial metabolism for phage replication. Rastogi et al [43], in their rat model for phage treatment of GI infection with E. coli, reported that phage-treated groups showed no damage or lesions in the GI tract, in comparison with untreated controls, which showed lymphomononuclear infiltration and edema. Hung et al [77], in their study for treatment of liver abscesses attributed to K. pneumoniae, measured circulating cytokines, as well as hepatic enzymes, and reported lower levels of TNF-α, IFN-γ, MCP-1, IL-10, and IL-6, as well as aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in phage-treated mice. Finally, in a murine model of P. aeruginosa keratitis, established by Fukuda et al [75], phage-treated mice had normal corneas, compared with a destroyed stromal structure and severe neutrophil infiltration of the corneas in untreated mice.
Following the promising results of phage treatment from the in vitro and preclinical in vivo data from animal models, bacteriophages have been tested in the clinical setting, particularly in patients whose infections do not respond to antibiotics. There have been several case reports of successful bacteriophage therapy in patients with different infections, and safety trials for phage targeting Gram-negative bacteria [82–84]. These reports focus on patient outcome and did not typically examine the kinetics of phage therapy as the urgency under which the bacteriophages were administered to patients precluded appropriate sampling of tissue or serum for bacteriophage titer. Likewise, histologic analysis and cytokine measurements were performed in only 2 of the 15 included reports.
Fifteen case reports and clinical trials for bacteriophage therapy were reviewed and are summarized in Table 5. Three studies involved pneumonia caused by P. aeruginosa [85–87], two reported the treatment of burn wound infections attributed to the same pathogen [82, 88], two dealt with bone infections, a left tibial lesion and a leg ulcer, both of which were polymicrobial infections. One case involved bacteriophage for treatment of necrotizing pancreatitis caused by A. baumannii [89], one applied phage for UTIs [90], another for treatment of acute diarrhea [91], 1 for treatment of P. aeruginosa septicemia [92], and one for treatment of hospital-acquired infections in an intensive care unit [93]. Another study reported the use of therapeutic bacteriophage against otitis caused by P. aeruginosa [94], and three studies were safety trials in healthy volunteers [83, 84,51].
Table 5. Human Studies
|
Reference |
Pathogen |
Animal Model |
Dose & Route of Administration |
Outcome |
Phage |
Histology & |
|
[82] |
P. aeruginosa |
Burn wound infection |
102 PFU, topically, once daily for 7 days (compared with the anticipated 106, phage titers were lowered after long storage) |
Phage-treated group had a longer time before 2 quadrant reduction in CFU was achieved (144h), compared with standard of care (47h). |
N/A |
N/A |
|
[85] |
P. aeruginosa |
Pneumonia in CF patient |
4x109 PFU, IV, every 6 hours, for 8 weeks |
The patient had no fever on day 7 and oxygen administration was reduced on day 8. The patient became ambulatory, was discharged from the hospital, and did not develop pneumonia for the 100-day follow up period; 9 months later she received a lung transplant |
N/A |
N/A |
|
[83] |
E. coli |
Safety trial - Healthy adult volunteers |
3x109 PFU (high dose) or 3x107 PFU (lower dose), orally |
No adverse effects in self-report, clinical evaluation and liver, kidney, and hematological examination |
64% of higher dose volunteers’ feces had phage at or below 3x107 PFU/mL only 30% of lower dose had phage (median phage concentration = 102 PFU) |
N/A |
|
[84] |
E. coli |
Safety trial - Healthy adult volunteers |
105 or 103 PFU/volunteer, orally |
No adverse effects were reported, except for a volunteer with a sore throat. Liver enzymes were normal in all volunteers |
105 PFU: 100% of volunteers had phage in their feces on day 3 (approximately 104 PFU/mL), which was retained until day 7 103 PFU: 50% prevalence in feces on days 2 and 3, phage was completely cleared by day 5 |
N/A |
|
[86] |
P. aeruginosa |
Pneumonia |
1st patient: 109 PFU, IV, every 6h nebulized every 12h, with concomitant antibiotics 2nd patient: 109 PFU, IV, every 12h |
1st patient: Improved within 29 days and had become ambulant by day 16. He presented back with pneumonia on day 46 (mucoid MDR P. aeruginosa) and re-started treatment. He was discharged from the hospital on day 150 after he improved and there was no active pneumonia for 3 months 2nd patient: Improved within 90 days, and she was discharged from the hospital. She came back with another infection that was non-MDR P. aeruginosa related. For the following 7 months, she was free of infection |
N/A |
N/A |
|
[87] |
P. aeruginosa |
VABP |
109 PFU, IV, and 4x109 PFU, nebulized, every 12 hours, with concomitant use of antibiotics |
Antibiotics alone did not improve the condition of the patient, but 3 days after phage initiation the patient improved dramatically, her %SaO2 increased and she was removed from intubation |
N/A |
N/A |
|
[88] |
P. aeruginosa |
Safety trial - Burn wound infection |
Single dose of 107 PFU, topically on half the wound (wound was divided into 2 sections: 1 received phage and the other standard treatment with amikacin, ceftazidime and meropenem) |
CFU counts were not significantly reduced after the use of phage |
N/A |
N/A |
|
[89] |
A. baumannii |
Necrotizing pancreatitis in diabetic patient |
4x109 PFU, initially through percutaneous catheters, after non-responsiveness, phage IV was also administered for a total of 18 weeks |
The patient awoke from coma and became conversant. The patient’s renal function (with an initial serum creatinine of 3.68 mg/dL) improved, and his general condition also improved gradually. |
5 min after administration, plasma levels of phage were 104 PFU/mL, and phage was almost completely eliminated from plasma within 60 min. After 6 hours, no phage was detected. |
N/A |
|
[90] |
P. aeruginosa and E. coli |
UTI |
107 – 109 PFU, through transurethral catheter, every 12h |
2-3 log10 reduction in CFU or complete eradication of the pathogen in some patients |
N/A |
N/A |
|
[91] |
E. coli |
Safety trial - Acute diarrhea - children |
Orally, 2 different cocktails, one of each group: Russian Microgen phage cocktail (M) and T4-like phage cocktail (T) |
No significant differences in duration of the diarrhea or clinical score of the patients or the quantitative factors (eg, diarrhea frequency, stool weight etc) were observed between the groups |
M group: highest phage titer of all groups and 100% phage-positive stool for 3 days. T group: Higher than placebo, as well as in the number of days with phage-positive stool rate |
Phage recipients had normal liver, kidney, and blood test results and did not have a positive Jarisch–Herxheimer reaction |
|
[92] |
P. aeruginosa |
Septicemia |
5x107 PFU, IV, every 6h and topically on wounds every 8h |
Blood cultures became negative, the fever dropped, and the patient’s kidney injury subsided |
N/A |
N/A |
|
[93] |
P. aeruginosa
K. pneumoniae A. baumannii |
Hospital- acquired infections |
5x108 PFU, orally, phage cocktail with 2 phages for each of the pathogens |
Before treatment, 79% of samples from patients’ endotracheal aspirate, blood, and urine were infected. After treatment, this rate dropped to 29% |
Phage was isolated from endotracheal aspirate, blood, feces, and urine 5 days after administration, showing that they cross the gastrointestinal epithelial barrier |
N/A |
|
[94] |
P. aeruginosa |
Otitis |
1.2x104 PFU, topically |
Visual Analog Scores were improved in patients of the treatment group. The phage-treated group showed improvement in inflammation, erythema, odor, and ulceration/ granulation/ polyps, compared to placebo group. CFU was reduced from 80-100% in phage-treated patients, compared with non-treated ones |
Mean PFU recovered from patients was 1.27x108, suggesting an amplification of approx. 200 times compared with the initial dose |
N/A |
|
[51] |
E. coli |
Safety trial - Healthy adult volunteers and healthy children |
7x106 or 7x105 PFU, orally |
No adverse effects in most of the patients. Abnormal eosinophil counts and liver function values were observed, but were present before the start of the treatment. One patient each reported a toothache, upset stomach, and ear infection. In general, no serious adverse effects were reported |
N/A |
N/A |
|
[95] |
P. aeruginosa and E. coli |
Leg ulcers – Safety trial |
Topically (dose is not mentioned) |
No difference in the healing rate between the 2 groups. They both had similar percentages of patients healed by week 12 and by week 24 |
N/A |
N/A |
|
[96] |
A. baumannii and K. pneumoniae |
Left tibial osteomyelitis |
5x107 PFU, IV over 35 minutes, for 5 days and then continued for an additional 6 days after Ab cultures were positive meropenem (2 grams tid), IV and colistin (4.5x106 units/bid), IV, were also administered |
Wound recovery started within few days of treatment and the wound eventually healed completely and patient’s pain disappeared; 8 months post-treatment, the patient did not show any re-infection with either organism |
N/A |
N/A |
Abbreviations: PFU: plaque forming unit; N/A: not applicable; IV: intravenous; CFU: colony forming unit; CF: cystic fibrosis; MDR: multidrug resistance; %SaO2: percent saturation of oxygen
All studies involving therapeutic use of bacteriophage, administered the cocktail intravenously at various dosing intervals of 6, 8, or 12 hours, or through the route that was consistent with the site of infection. Thus, wound infections were treated with topical administration of bacteriophage, in UTIs through trans-urethral catheters, and in pneumonias via the intravenous or nebulized routes. In all therapeutic applications of bacteriophage, the dose ranged in the order of 107 – 109 PFU, with the exception of the study by Jault et al [82], where the administered dose was 102 PFU, due to a marked reduction in the phage titer after long storage.
In all but 1 reported case of therapeutic application of phage, the patients recovered from a critical condition without re-infection for follow-up periods that were as long as 9 months. Only 2 of the therapeutic application studies examined the kinetics of phage. In the study by Schooley et al [89], phage concentration in plasma was reduced to less than half the initial dose within 5 minutes and was almost completely eliminated within 60 minutes. After 6 hours, no phage could be found in the plasma. Aleshkin et al [93] were able to isolate phage from the endotracheal aspirate, blood, feces, and urine for up to 5 days after administration through an orogastric tube. These observations demonstrate that bacteriophage can translocate across the gastrointestinal epithelium. None of these studies examined the histological and immunological aspects of phage therapy in humans.
Both healthy volunteers and sick patients participated in safety trials of bacteriophage therapy. The doses used in those cases had a wider range, between 103 and 109 PFU, and were administered either orally or topically, as in the case of burn wound infections [88] or leg ulcers [95]. These studies mainly examined the safety of phage administration. Evaluation of the safety of bacteriophage was determined by clinical assessment, as well as by biochemical parameters, including hepatic and renal function, and hematological parameters. Phage therapy was well tolerated with few mild self-limiting adverse events reported [51, 84].
During phage therapy of a patient with disseminated resistant A. baumannii infection, emergence of resistance occurred to the first bacteriophage cocktail. In order to overcome resistance, 2 consecutive phage cocktails were administered during the course of the patient’s extended treatment [89]. Since emergence of resistance of infecting bacteria to therapeutic phages is a potential limitation to phage therapy, rational strategies are needed to anticipate and prevent these events.
The 3 studies which reported phage kinetic data [83, 84, 91] demonstrated that after oral or IV administration, phage persisted in all patients’ feces for 3 days and for as much as 7 days in many cases. In terms of immune response evaluation, only Sarker et al [91] reported that the children in their study did not have a positive Jarisch-Herxheimer-like reaction, which would be indicative of endotoxin release after the lysis of bacterial cells. No other studies in humans have examined the histology or cytokine immune response of the patients.
Current studies thus far have demonstrated that intravenously administered phage therapies for Gram-negative bacillary infections, have minimal adverse effects, thus avoiding possible toxicity to seriously ill patients with multidrug resistant Gram-negative bacterial infections, something that could complicate treatment. Among studies that examined safety of phage application, the following adverse events were reported: A transient hypertension in a patient with A. baumannii infection, which did not require vasopressors, and an increase in blood pressure in a different patient with an A. baumannii infection, which lasted for approximately a week but was resolved after temporary discontinuation of the phage administration. In a patient with a P. aeruginosa infection, a 38.5°C fever was observed, as well as chills, on the third day of phage therapy. In the most serious side effect observed so far, concerning a 2-year-old child with a P. aeruginosa infection, phage therapy was withheld due to anaphylaxis-related decompensation, which was attributed to progressive heart failure. Finally, 1 patient reported mild headaches, and another patient reported transient ear pain during phage therapy. In most cases, those side effects are attributed to the endolysin release after killing of the bacteria by the phage. These side effects may be reduced or completely minimized when using a lysis-deficient phage, as was the case in the study by Matsuda et al. Independent of the route of administration, phages are a safe therapeutic intervention [83, 84]. In the studies that monitored potential side effects with laboratory tests, such as in a randomized trial for oral phage therapy of acute diarrhea in children [91], phage recipients had normal liver, kidney, and blood test results and did not have a positive Jarisch-Herxheimer-like reaction.
As the result of being highly targeted to a specific host bacterial pathogen, bacteriophage therapy is less likely than conventional small molecule antimicrobial agents to significantly disturb the normal gastrointestinal microbiota. By comparison, broad spectrum antimicrobial agents that are currently used to treat multidrug resistant Gram-negative bacterial infections, inevitably lead to disruption of the gastrointestinal microbiota and potentially lead to antibiotic-associated diarrhea, including Clostridium difficile-associated diarrhea.
In terms of the pharmacological aspects of bacteriophage therapy, phages and conventional antibiotics share some common properties that govern their application. Both bacteriophages and antimicrobials display either concentration-dependent or time-dependent killing. In certain infections, depending on the organism and type of infection, phage treatment may be concentration-dependent and similar to the Cmax/MIC ratio in conventional antibiotics, such as aminoglycosides or fluoroquinolones. In this case, the multiplicity of infection or MOI (ie, the ratio of phage particles divided by the pathogen load at the time of infection) may play an important role in the outcome of the treatment. This is similar to the effect of the peak plasma concentration (Cmax) of an antibiotic, divided by its minimal inhibitory concentration (MIC), as observed in concentration-dependent antibiotics. Contrary to that, time-dependent activity (defined as the time spent above the MIC in the case of conventional antibiotics) means that dosing intervals should be set in such a way that phage concentration is retained above the necessary MOI long enough to achieve killing of the organism (Figure 2). Deriving this is complicated by ongoing phage replication in the target pathogen during treatment, and it is likely that this replication rate varies by compartment and even by microenvironment. Dose optimization may be conducted by serial quantification of bacteriophages in blood and tissue, as demonstrated. Alternatively, a more common approach for dose optimization of bacteriophage is conducted in vivo through empirical evaluation of dosages and dosing intervals.
Another similarity between bacteriophages and antibiotics that this review highlights, is the need for pharmaceutical formulations which will assure that the antimicrobial will be delivered to the infected target. For example, providing an enteric film coating, which will protect an antibiotic from degradation in the stomach and help release it in the small intestine, can also be applied to bacteriophage [78, 95]. Likewise, coating of the phage with liposomes (Figure 3), as mentioned in several studies reported herein, can release the phage in a more targeted way, reducing the phage concentrations needed for activity. Such is the case in the studies by Singla et al [47] and Chadha et al [57], which showed that liposome entrapment can improve therapeutic outcomes even when the phage concentration and MOI are much lower than those used in non-liposome entrapped therapies.
In many of the studies mentioned above, phage kinetics with regard to bacterial host encountering was evaluated. More specifically, these studies demonstrated that when phage encountered host bacteria, they replicated and therefore remained in circulation and in tissues longer than if there was no bacterial host. To optimize the phage-host interaction would require study of the optimal initial dose to combat a specific bacterial CFU, while also investigating the increase of MOI as bacteriophage replicate in host bacterium they encounter. More extensive studies that address these pharmacokinetic properties of bacteriophage and multidrug resistant bacteria are necessary to make further advances in the field of phage therapeutics.
Finally, another distinctive property of phage therapy may be the manipulation of phage dose based on host membrane structure. As shown by Bull et al [70], specificity of the phage to a bacterial surface feature that was also required for virulence greatly enhanced the outcome of treatment. This finding suggests that low PFU doses, and thus less phage product, may be as effective as high PFU doses in well-tailored phage-therapy treatments.
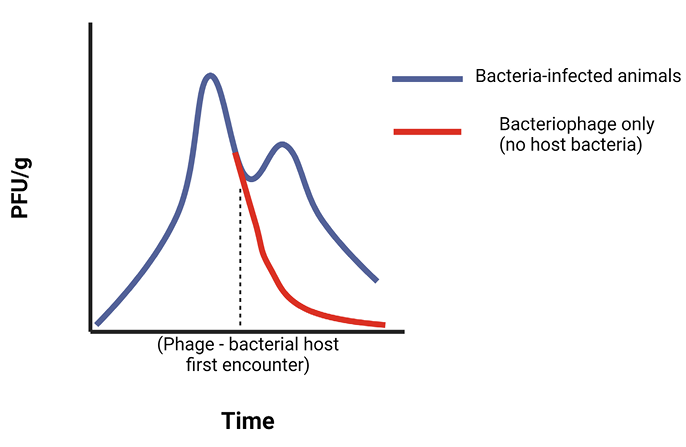
Figure 2A. Conceptual model of the effect of bacterial infection on phage (Φ) kinetics in blood and tissues. When phage encounter their bacterial hosts (bh) in infected animals, they replicate, and therefore require a longer time to be cleared from the system, in comparison to that of uninfected animals, where there are no bacteria that phage can infect. This self-replication property extends their therapeutic effect, as it leads to higher titers in tissues and blood, potentially leading to a reduced need for many doses, when compared with conventional antibiotics.
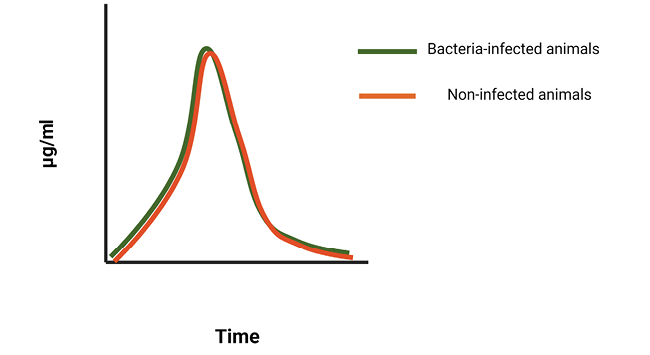
Figure 2B. Conceptual model of the kinetics of conventional antibiotics in blood and tissues. In comparison to bacteriophages, conventional antibiotics cannot replicate and are cleared from the system within the same time, regardless of whether or not a bacterial infection is present in animals.
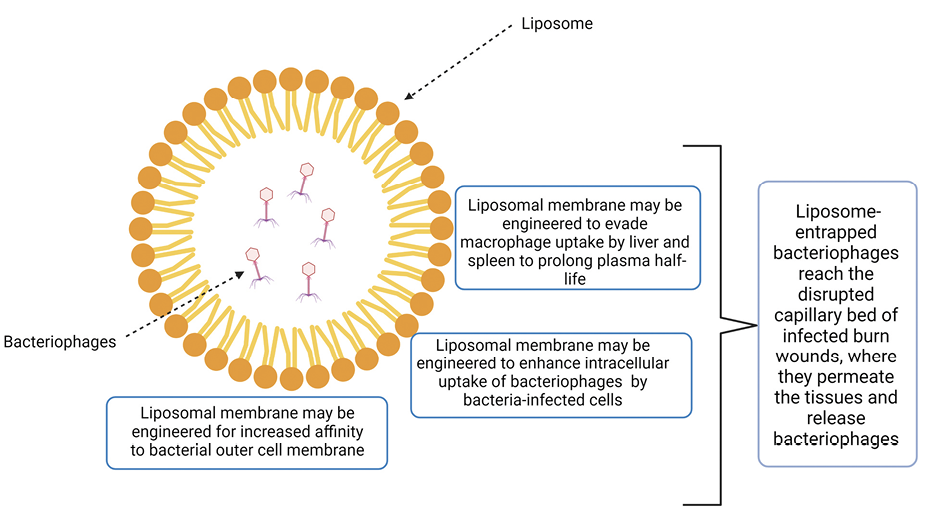
Figure 3. Possible mechanisms of enhanced antimicrobial activity of liposome-entrapped bacteriophages (LEBs). The chemical composition and structure of liposomes of LEBs may be engineered for polarity, size, and lipophilicity to evade macrophage uptake by liver and spleen to prolong the plasma half-life of bacteriophages, to enhance intracellular uptake of LEBs by bacteria-infected cells, and to increase affinity to bacterial outer cell membranes. Moreover, LEBs reach the disrupted capillary bed of infected burn wounds where they permeate the tissues and release bacteriophages.
While animal studies and individual case reports have indicated that phage therapy can be effective for the treatment of bacterial infections, the path ahead presents multiple challenges. Commercial development of therapeutic phages is an ongoing process: phage products are already available for food safety applications [72], for the control of plant pathogens, for use in aquaculture, and as nutritional supplements. These areas have been more attractive for initial commercialization as the regulatory requirements for marketing in these sectors are generally much simpler than for medicines. As of this writing, there are 19 clinical trials registered on clinicaltrials.gov for the evaluation of phage-based therapeutics to treat a variety of infection types; 7 of these clinical trials are recruiting patients.
The high specificity of phages, seen as an advantage for avoiding collateral damage to non-pathogenic flora, is also a challenge for the development of phages as broadly applicable therapeutics. Because a given phage will only infect a subset of strains within a bacterial species, multiple phages will be needed to adequately cover the most clinically relevant strains of a pathogen. Given the propensity for bacterial resistance to individual bacteriophages, optimized clinical application of phages against MDR Gram-negative bacilli will likely require the use of phage mixtures, or “cocktails”. Cocktail phages may be chosen based on their host range across multiple pathogen strains, their ability to forestall the emergence of resistance, their favorable PK/PD characteristics, and their ability to minimize the probability of emergence of resistance within bacterial populations causing infection [43]. The desired properties may be obtained by identifying and combining naturally-occurring phages, or by engineering phages to possess the desired characteristics [48]. Many aspects of basic phage biology remain to be discovered to fully exploit their therapeutic potential, but phage engineering is also attractive because it provides much stronger intellectual property (IP) protection for the final product, a significant concern in commercial drug development.
Treatment of Gram-negative bacterial infections with phage may result in the rapid release of lipopolysaccharide with subsequent pro-inflammatory and hemodynamic deterioration in the infected host. This deleterious effect may be circumvented by lysis-deficient (LyD) phage. Further development of such phage cocktails may be a safer alternative to the use of lytic phages.
Two broad models of phage development have been proposed [97]. The first model resembles a more traditional, fixed-composition product designed to treat a specific pathogen or condition. Such a product would have a clearly defined identity and would move through a regulatory approval process that resembles that used for other small-molecule drugs or biologicals. This approach has the advantages of clearer pathways for both regulatory approval and IP protection. The disadvantage of this approach is the static nature of the product, which may not be able to treat all pathogen strains circulating in a patient population and which cannot adapt to the emergence of new strains that are not susceptible to the phages in the product. This disadvantage may be allayed in part by the implementation of a more flexible regulatory framework that would allow for periodic updating or versioning of the product to adapt to current needs without the need for completely new sets of preclinical and clinical studies. The second proposed model is a more flexible, personalized medicine approach in which a mixture of therapeutic phages is formulated on-demand based on the phage sensitivity of an individual patient’s pathogen isolate. This model has the advantages of adaptability to individual patient needs and is thus less likely to be hampered by individual strain variability, but details on how to scale this approach to a level that would make it broadly available are yet to be determined. Additionally, the regulatory pathway for this approach is much less defined, as there is no single fixed-definition molecular entity to be approved. The magistral preparation approach used in some European Union jurisdictions may be compatible with phage personalized medicine approaches [44], but the approval process for this implementation in other jurisdictions remains to be clarified. Magistral preparation refers to the formulation of a medicinal product by a pharmacy that fills a prescription for a specific patient and not requiring marketing or manufacturing licensing or authorization.
Another approach of harnessing the potential power of phage therapy is to administer modified lysins for treatment of life-threatening infections. Following successful preclinical investigations, a phase 2 clinical trial of lysin plus standard antibacterial therapy was found to be more active than standard antibacterial therapy alone in treatment of MRSA bacteremia and right-sided endocarditis [98]. These results led to a phase 3 clinical trial, which unfortunately was closed for lack of feasibility. Further preclinical studies are warranted to optimize the activity of lysin in MRSA infections as a guide to future clinical trials. Current and future preclinical studies are underway to investigate the antimicrobial activity of lysins and amurins for treatment of infections caused by P. aeruginosa and A. baumannii [49].
Finally, economic issues that affect anti-infective development will also affect phage therapy. The stand-out application of phage therapy is the treatment of multidrug-resistant or pan-resistant organisms, which are not responsive to treatment by conventional antibiotics. While the threat of antimicrobial resistant (AMR) bacteria continues to grow, these infections represent only a portion of all cases, and thus the current market for novel therapeutics against these infections is not as large as the market for antibacterial agents as a whole. That antibacterial development has been seen as economically unattractive poses a challenge for conventional commercial investment [46]. However, as the frequency of AMR infections continues to rise, it would seem prudent to invest in new antibacterial strategies in order that they can be in place for future generations.
The global emergence of multidrug-resistant Gram-negative bacterial infections threatens the limited antimicrobial armamentarium. Bacteriophage therapy offers a compelling adjunct to development of new antimicrobial agents to meet this global challenge. Depending upon the type of infection, the level of resistance by the target bacteria, and efficacy of available antibiotics, clinical trials of phage therapy may be designed as adjunctive therapy or monotherapy. Animal model systems can be used as powerful tools for the development of new bacteriophage therapies, as well as for designing and lowering the risk of clinical trials. Bacteriophage therapy for human use has currently been limited to a small number of individual cases, but these treatments have demonstrated the proof-of-concept that phages can be used to combat life-threatening antimicrobial resistant bacterial infections. Going forward, a focus on pre-clinical pharmacokinetics and pharmacodynamic optimization of bacteriophage therapy against multidrug-resistant bacterial infections, using quantifiable endpoints of residual tissue bacterial burden, survival, and pro-inflammatory cytokine profiles, will be critical in obtaining a better understanding of bacteriophage efficacy in a clinical setting. Additionally, the development of highly predictive animal model systems, well-designed clinical trials, and reliable sources of produced bacteriophage will be essential in further improvement of this promising technology in combatting multidrug-resistant Gram-negative infections.
TJW has received grants for experimental and clinical antimicrobial pharmacology, therapeutics, and diagnostics to his institution from Allergan, Amplyx, Astellas, Lediant, Merck, Medicines Company, Scynexis, Shionogi, T2 Biosystems, Tetraphase, and Viosera; and served as consultant to Amplyx, Astellas, Allergan, ContraFect, Gilead, Karyopharm, Leadiant, Medicines Company, Merck, Methylgene, Partner Therapeutics, Pfizer, Scynexis, Shionogi, Statera, and T2 Biosystems
This research was supported in part by R21/R33 AI121689-01 (JG, TJW, JZ, and JM-P), the Hellenic-American Scholarship-Mentorship (TJW and JZ) by a Scholar award from the Save Our Sick Kids Foundation (TJW)
1. Dhingra S, Rahman NAA, Peile E, Rahman M, Sartelli M, Hassali MA, Islam T, Islam S, Haque M. Microbial Resistance Movements: An Overview of Global Public Health Threats Posed by Antimicrobial Resistance, and How Best to Counter. Front Public Health. 2020;8:535668. doi: 10.3389/fpubh.2020.535668. PubMed PMID: 33251170; PMCID: PMC7672122.
2. Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74(3):417-33. doi: 10.1128/MMBR.00016-10. PubMed PMID: 20805405; PMCID: PMC2937522.
3. Exner M, Bhattacharya S, Christiansen B, Gebel J, Goroncy-Bermes P, Hartemann P, Heeg P, Ilschner C, Kramer A, Larson E, Merkens W, Mielke M, Oltmanns P, Ross B, Rotter M, Schmithausen RM, Sonntag HG, Trautmann M. Antibiotic resistance: What is so special about multidrug-resistant Gram-negative bacteria? GMS Hyg Infect Control. 2017;12:Doc05. doi: 10.3205/dgkh000290. PubMed PMID: 28451516; PMCID: PMC5388835.
4. World Health O. Antibiotic resistance: multi-country public awareness survey. Geneva: World Health Organization; 2015.
5. Bonomo RA, Burd EM, Conly J, Limbago BM, Poirel L, Segre JA, Westblade LF. Carbapenemase-Producing Organisms: A Global Scourge. Clin Infect Dis. 2018;66(8):1290-7. doi: 10.1093/cid/cix893. PubMed PMID: 29165604; PMCID: PMC5884739.
6. Reygaert WC. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018;4(3):482-501. doi: 10.3934/microbiol.2018.3.482. PubMed PMID: 31294229; PMCID: PMC6604941.
7. Karakonstantis S, Rousaki M, Kritsotakis EI. Cefiderocol: Systematic Review of Mechanisms of Resistance, Heteroresistance and In Vivo Emergence of Resistance. Antibiotics (Basel). 2022;11(6). doi: 10.3390/antibiotics11060723. PubMed PMID: 35740130; PMCID: PMC9220290.
8. Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18(3):268-81. doi: 10.1111/j.1469-0691.2011.03570.x. PubMed PMID: 21793988.
9. Mohapatra DP, Debata NK, Singh SK. Extensively drug-resistant and pandrug-resistant Gram-negative bacteria in a tertiary-care hospital in Eastern India: A 4-year retrospective study. J Glob Antimicrob Resist. 2018;15:246-9. doi: 10.1016/j.jgar.2018.08.010. PubMed PMID: 30144638.
10. Alanis AJ. Resistance to antibiotics: are we in the post-antibiotic era? Arch Med Res. 2005;36(6):697-705. doi: 10.1016/j.arcmed.2005.06.009. PubMed PMID: 16216651.
11. Twort FW. An Investigation on the Nature of Ultra-Microscopic Viruses. Lancet. 1915;186(4814):1241-3. doi: 10.1016/s0140-6736(01)20383-3.
12. Keen EC. A century of phage research: bacteriophages and the shaping of modern biology. Bioessays. 2015;37(1):6-9. doi: 10.1002/bies.201400152. PubMed PMID: 25521633; PMCID: PMC4418462.
13. Fernandes S, Sao-Jose C. Enzymes and Mechanisms Employed by Tailed Bacteriophages to Breach the Bacterial Cell Barriers. Viruses. 2018;10(8). doi: 10.3390/v10080396. PubMed PMID: 30060520; PMCID: PMC6116005.
14. Jasim HN, Hafidh RR, Abdulamir AS. Formation of therapeutic phage cocktail and endolysin to highly multi-drug resistant Acinetobacter baumannii: in vitro and in vivo study. Iran J Basic Med Sci. 2018;21(11):1100-8. doi: 10.22038/IJBMS.2018.27307.6665. PubMed PMID: 30483382; PMCID: PMC6251394.
15. Wang C, Li P, Niu W, Yuan X, Liu H, Huang Y, An X, Fan H, Zhangxiang L, Mi L, Zheng J, Liu Y, Tong Y, Mi Z, Bai C. Protective and therapeutic application of the depolymerase derived from a novel KN1 genotype of Klebsiella pneumoniae bacteriophage in mice. Res Microbiol. 2019;170(3):156-64. doi: 10.1016/j.resmic.2019.01.003. PubMed PMID: 30716390.
16. Lin H, Paff ML, Molineux IJ, Bull JJ. Therapeutic Application of Phage Capsule Depolymerases against K1, K5, and K30 Capsulated E. coli in Mice. Front Microbiol. 2017;8:2257. doi: 10.3389/fmicb.2017.02257. PubMed PMID: 29201019; PMCID: PMC5696595.
17. Mushtaq N, Redpath MB, Luzio JP, Taylor PW. Treatment of experimental Escherichia coli infection with recombinant bacteriophage-derived capsule depolymerase. J Antimicrob Chemother.2005;56(1):160-5. doi: 10.1093/jac/dki177. PubMed PMID: 15914489.
18. Gill JJ, Hyman P. Phage choice, isolation, and preparation for phage therapy. Curr Pharm Biotechnol. 2010;11(1):2-14. doi: 10.2174/138920110790725311. PubMed PMID: 20214604.
19. El Haddad L, Harb CP, Gebara MA, Stibich MA, Chemaly RF. A Systematic and Critical Review of Bacteriophage Therapy Against Multidrug-resistant ESKAPE Organisms in Humans. Clin Infect Dis. 2019;69(1):167-78. doi: 10.1093/cid/ciy947. PubMed PMID: 30395179.
20. Kortright KE, Chan BK, Koff JL, Turner PE. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe. 2019;25(2):219-32. doi: 10.1016/j.chom.2019.01.014. PubMed PMID: 30763536.
21. Bielen K, s Jongers B, Malhotra-Kumar S, Jorens PG, Goossens H, Kumar-Singh S. Animal models of hospital-acquired pneumonia: current practices and future perspectives. Ann Transl Med. 2017;5(6):132. doi: 10.21037/atm.2017.03.72. PubMed PMID: 28462212; PMCID: PMC5395468.
22. Waack U, Weinstein EA, Farley JJ. Assessing Animal Models of Bacterial Pneumonia Used in Investigational New Drug Applications for the Treatment of Bacterial Pneumonia. Antimicrob Agents Chemother. 2020;64(5). doi: 10.1128/AAC.02242-19. PubMed PMID: 32122895; PMCID: PMC7179594.
23. Hua Y, Luo T, Yang Y, Dong D, Wang R, Wang Y, Xu M, Guo X, Hu F, He P. Phage Therapy as a Promising New Treatment for Lung Infection Caused by Carbapenem-Resistant Acinetobacter baumannii in Mice. Front Microbiol. 2017;8:2659. doi: 10.3389/fmicb.2017.02659. PubMed PMID: 29375524; PMCID: PMC5767256.
24. Dufour N, Clermont O, La Combe B, Messika J, Dion S, Khanna V, Denamur E, Ricard JD, Debarbieux L, ColoColi g. Bacteriophage LM33_P1, a fast-acting weapon against the pandemic ST131-O25b:H4 Escherichia coli clonal complex. J Antimicrob Chemother.2016;71(11):3072-80. doi: 10.1093/jac/dkw253. PubMed PMID: 27387322.
25. Raz A, Serrano A, Hernandez A, Euler CW, Fischetti VA. Isolation of Phage Lysins That Effectively Kill Pseudomonas aeruginosa in Mouse Models of Lung and Skin Infection. Antimicrob Agents Chemother. 2019;63(7). doi: 10.1128/AAC.00024-19. PubMed PMID: 31010858; PMCID: PMC6591642.
26. Alemayehu D, Casey PG, McAuliffe O, Guinane CM, Martin JG, Shanahan F, Coffey A, Ross RP, Hill C. Bacteriophages phiMR299-2 and phiNH-4 can eliminate Pseudomonas aeruginosa in the murine lung and on cystic fibrosis lung airway cells. mBio. 2012;3(2):e00029-12. doi: 10.1128/mBio.00029-12. PubMed PMID: 22396480; PMCID: PMC3302570.
27. Waters EM, Neill DR, Kaman B, Sahota JS, Clokie MRJ, Winstanley C, Kadioglu A. Phage therapy is highly effective against chronic lung infections with Pseudomonas aeruginosa. Thorax. 2017;72(7):666-7. doi: 10.1136/thoraxjnl-2016-209265. PubMed PMID: 28265031; PMCID: PMC5520275.
28. Cao Z, Zhang J, Niu YD, Cui N, Ma Y, Cao F, Jin L, Li Z, Xu Y. Isolation and characterization of a “phiKMV-like” bacteriophage and its therapeutic effect on mink hemorrhagic pneumonia. PLoS One. 2015;10(1):e0116571. doi: 10.1371/journal.pone.0116571. PubMed PMID: 25615639; PMCID: PMC4304800.
29. Roach DR, Leung CY, Henry M, Morello E, Singh D, Di Santo JP, Weitz JS, Debarbieux L. Synergy between the Host Immune System and Bacteriophage Is Essential for Successful Phage Therapy against an Acute Respiratory Pathogen. Cell Host Microbe. 2017;22(1):38-47 e4. doi: 10.1016/j.chom.2017.06.018. PubMed PMID: 28704651.
30. Huff WE, Huff GR, Rath NC, Balog JM, Donoghue AM. Evaluation of aerosol spray and intramuscular injection of bacteriophage to treat an Escherichia coli respiratory infection. Poult Sci. 2003;82(7):1108-12. doi: 10.1093/ps/82.7.1108. PubMed PMID: 12872966.
31. Christaki E, Giamarellos-Bourboulis EJ. The complex pathogenesis of bacteremia: from antimicrobial clearance mechanisms to the genetic background of the host. Virulence. 2014;5(1):57-65. doi: 10.4161/viru.26514. PubMed PMID: 24067507; PMCID: PMC3916384.
32. Yin S, Huang G, Zhang Y, Jiang B, Yang Z, Dong Z, You B, Yuan Z, Hu F, Zhao Y, Peng Y. Phage Abp1 Rescues Human Cells and Mice from Infection by Pan-Drug Resistant Acinetobacter Baumannii. Cell Physiol Biochem. 2017;44(6):2337-45. doi: 10.1159/000486117. PubMed PMID: 29258062.
33. Schneider G, Szentes N, Horvath M, Dorn A, Cox A, Nagy G, Doffkay Z, Maroti G, Rakhely G, Kovacs T. Kinetics of Targeted Phage Rescue in a Mouse Model of Systemic Escherichia coli K1. Biomed Res Int. 2018;2018:7569645. doi: 10.1155/2018/7569645. PubMed PMID: 30105246; PMCID: PMC6076946.
34. Horvath M, Kovacs T, Koderivalappil S, Abraham H, Rakhely G, Schneider G. Identification of a newly isolated lytic bacteriophage against K24 capsular type, carbapenem resistant Klebsiella pneumoniae isolates. Sci Rep. 2020;10(1):5891. doi: 10.1038/s41598-020-62691-8. PubMed PMID: 32246126; PMCID: PMC7125228.
35. Kaabi SAG, Musafer HK. An experimental mouse model for phage therapy of bacterial pathogens causing bacteremia. Microb Pathog. 2019;137:103770. doi: 10.1016/j.micpath.2019.103770. PubMed PMID: 31586662.
36. Alvi IA, Asif M, Tabassum R, Aslam R, Abbas Z, Rehman SU. RLP, a bacteriophage of the family Podoviridae, rescues mice from bacteremia caused by multi-drug-resistant Pseudomonas aeruginosa. Arch Virol. 2020;165(6):1289-97. doi: 10.1007/s00705-020-04601-x. PubMed PMID: 32246283.
37. Lin YW, Chang RY, Rao GG, Jermain B, Han ML, Zhao JX, Chen K, Wang JP, Barr JJ, Schooley RT, Kutter E, Chan HK, Li J. Pharmacokinetics/pharmacodynamics of antipseudomonal bacteriophage therapy in rats: a proof-of-concept study. Clin Microbiol Infect. 2020;26(9):1229-35. doi: 10.1016/j.cmi.2020.04.039. PubMed PMID: 32387436.
38. Heselpoth RD, Euler CW, Schuch R, Fischetti VA. Lysocins: Bioengineered Antimicrobials That Deliver Lysins across the Outer Membrane of Gram-Negative Bacteria. Antimicrob Agents Chemother. 2019;63(6). doi: 10.1128/AAC.00342-19. PubMed PMID: 30962344; PMCID: PMC6535517.
39. Lood R, Winer BY, Pelzek AJ, Diez-Martinez R, Thandar M, Euler CW, Schuch R, Fischetti VA. Novel phage lysin capable of killing the multidrug-resistant gram-negative bacterium Acinetobacter baumannii in a mouse bacteremia model. Antimicrob Agents Chemother. 2015;59(4):1983-91. doi: 10.1128/AAC.04641-14. PubMed PMID: 25605353; PMCID: PMC4356752.
40. Yehl K, Lemire S, Yang AC, Ando H, Mimee M, Torres MT, de la Fuente-Nunez C, Lu TK. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell. 2019;179(2):459-69 e9. doi: 10.1016/j.cell.2019.09.015. PubMed PMID: 31585083; PMCID: PMC6924272.
41. Chadha P, Katare OP, Chhibber S. In vivo efficacy of single phage versus phage cocktail in resolving burn wound infection in BALB/c mice. Microb Pathog. 2016;99:68-77. doi: 10.1016/j.micpath.2016.08.001. PubMed PMID: 27498362.
42. Nishikawa H, Yasuda M, Uchiyama J, Rashel M, Maeda Y, Takemura I, Sugihara S, Ujihara T, Shimizu Y, Shuin T, Matsuzaki S. T-even-related bacteriophages as candidates for treatment of Escherichia coli urinary tract infections. Arch Virol. 2008;153(3):507-15. doi: 10.1007/s00705-007-0031-4. PubMed PMID: 18188500.
43. Rastogi V, Yadav P, Verma A, Pandit JK. Ex vivo and in vivo evaluation of microemulsion based transdermal delivery of E. coli specific T4 bacteriophage: A rationale approach to treat bacterial infection. Eur J Pharm Sci. 2017;107:168-82. doi: 10.1016/j.ejps.2017.07.014. PubMed PMID: 28711712.
44. Verbeken G, Pirnay JP. European regulatory aspects of phage therapy: magistral phage preparations. Curr Opin Virol. 2022;52:24-9. doi: 10.1016/j.coviro.2021.11.005. PubMed PMID: 34801778.
45. Smith HW, Huggins MB. Successful treatment of experimental Escherichia coli infections in mice using phage: its general superiority over antibiotics. J Gen Microbiol. 1982;128(2):307-18. doi: 10.1099/00221287-128-2-307. PubMed PMID: 7042903.
46. Bull JJ, Levin BR, DeRouin T, Walker N, Bloch CA. Dynamics of success and failure in phage and antibiotic therapy in experimental infections. BMC Microbiol. 2002;2:35. doi: 10.1186/1471-2180-2-35. PubMed PMID: 12453306; PMCID: PMC138797.
47. Singla S, Harjai K, Katare OP, Chhibber S. Bacteriophage-loaded nanostructured lipid carrier: improved pharmacokinetics mediates effective resolution of Klebsiella pneumoniae-induced lobar pneumonia. J Infect Dis. 2015;212(2):325-34. doi: 10.1093/infdis/jiv029. PubMed PMID: 25605867.
48. Gibb B, Hyman P, Schneider CL. The Many Applications of Engineered Bacteriophages-An Overview. Pharmaceuticals (Basel). 2021;14(7). doi: 10.3390/ph14070634. PubMed PMID: 34208847; PMCID: PMC8308837.
49. Schuch R, Cassino C, Vila-Farres X. Direct Lytic Agents: Novel, Rapidly Acting Potential Antimicrobial Treatment Modalities for Systemic Use in the Era of Rising Antibiotic Resistance. Front Microbiol. 2022;13:841905. doi: 10.3389/fmicb.2022.841905. PubMed PMID: 35308352; PMCID: PMC8928733.
50. Ardal C, Balasegaram M, Laxminarayan R, McAdams D, Outterson K, Rex JH, Sumpradit N. Antibiotic development - economic, regulatory and societal challenges. Nat Rev Microbiol. 2020;18(5):267-74. doi: 10.1038/s41579-019-0293-3. PubMed PMID: 31745330.
51. McCallin S, Alam Sarker S, Barretto C, Sultana S, Berger B, Huq S, Krause L, Bibiloni R, Schmitt B, Reuteler G, Brussow H. Safety analysis of a Russian phage cocktail: from metagenomic analysis to oral application in healthy human subjects. Virology. 2013;443(2):187-96. doi: 10.1016/j.virol.2013.05.022. PubMed PMID: 23755967.
52. Cha K, Oh HK, Jang JY, Jo Y, Kim WK, Ha GU, Ko KS, Myung H. Characterization of Two Novel Bacteriophages Infecting Multidrug-Resistant (MDR) Acinetobacter baumannii and Evaluation of Their Therapeutic Efficacy in Vivo. Front Microbiol. 2018;9:696. doi: 10.3389/fmicb.2018.00696. PubMed PMID: 29755420; PMCID: PMC5932359.
53. Wang Y, Mi Z, Niu W, An X, Yuan X, Liu H, Li P, Liu Y, Feng Y, Huang Y, Zhang X, Zhang Z, Fan H, Peng F, Tong Y, Bai C. Intranasal treatment with bacteriophage rescues mice from Acinetobacter baumannii-mediated pneumonia. Future Microbiol. 2016;11:631-41. doi: 10.2217/fmb.16.11. PubMed PMID: 26925593.
54. Soleimani Sasani M, Eftekhar F. Potential of a Bacteriophage Isolated from Wastewater in Treatment of Lobar Pneumonia Infection Induced by Klebsiella pneumoniae in Mice. Curr Microbiol. 2020;77(10):2650-5. doi: 10.1007/s00284-020-02041-z. PubMed PMID: 32451685.
55. Jeon J, Yong D. Two Novel Bacteriophages Improve Survival in Galleria mellonella Infection and Mouse Acute Pneumonia Models Infected with Extensively Drug-Resistant Pseudomonas aeruginosa. Appl Environ Microbiol. 2019;85(9). doi: 10.1128/AEM.02900-18. PubMed PMID: 30824445; PMCID: PMC6495756.
56. Jeon J, Park JH, Yong D. Efficacy of bacteriophage treatment against carbapenem-resistant Acinetobacter baumannii in Galleria mellonella larvae and a mouse model of acute pneumonia. BMC Microbiol. 2019;19(1):70. doi: 10.1186/s12866-019-1443-5. PubMed PMID: 30940074; PMCID: PMC6444642.
57. Chadha P, Katare OP, Chhibber S. Liposome loaded phage cocktail: Enhanced therapeutic potential in resolving Klebsiella pneumoniae mediated burn wound infections. Burns. 2017;43(7):1532-43. doi: 10.1016/j.burns.2017.03.029. PubMed PMID: 28502784.
58. Rahmani R, Zarrini G, Sheikhzadeh F, Aghamohammadzadeh N. Effective Phages as Green Antimicrobial Agents Against Antibiotic-Resistant Hospital Escherichia coli. Jundishapur J Microbiol. 2015;8(2):e17744. doi: 10.5812/jjm.17744. PubMed PMID: 25834712; PMCID: PMC4377167.
59. Kang HW, Kim JW, Jung TS, Woo GJ. wksl3, a New biocontrol agent for Salmonella enterica serovars enteritidis and typhimurium in foods: characterization, application, sequence analysis, and oral acute toxicity study. Appl Environ Microbiol. 2013;79(6):1956-68. doi: 10.1128/AEM.02793-12. PubMed PMID: 23335772; PMCID: PMC3592225.
60. Dufour N, Debarbieux L, Fromentin M, Ricard JD. Treatment of Highly Virulent Extraintestinal Pathogenic Escherichia coli Pneumonia With Bacteriophages. Crit Care Med. 2015;43(6):e190-8. doi: 10.1097/CCM.0000000000000968. PubMed PMID: 25803649.
61. Henry M, Lavigne R, Debarbieux L. Predicting in vivo efficacy of therapeutic bacteriophages used to treat pulmonary infections. Antimicrob Agents Chemother. 2013;57(12):5961-8. doi: 10.1128/AAC.01596-13. PubMed PMID: 24041900; PMCID: PMC3837875.
62. Chang RYK, Chen K, Wang J, Wallin M, Britton W, Morales S, Kutter E, Li J, Chan HK. Proof-of-Principle Study in a Murine Lung Infection Model of Antipseudomonal Activity of Phage PEV20 in a Dry-Powder Formulation. Antimicrob Agents Chemother. 2018;62(2). doi: 10.1128/AAC.01714-17. PubMed PMID: 29158280; PMCID: PMC5786808.
63. Chung IY, Sim N, Cho YH. Antibacterial efficacy of temperate phage-mediated inhibition of bacterial group motilities. Antimicrob Agents Chemother. 2012;56(11):5612-7. doi: 10.1128/AAC.00504-12. PubMed PMID: 22908158; PMCID: PMC3486593.
64. Tiwari BR, Kim S, Rahman M, Kim J. Antibacterial efficacy of lytic Pseudomonas bacteriophage in normal and neutropenic mice models. J Microbiol. 2011;49(6):994-9. doi: 10.1007/s12275-011-1512-4. PubMed PMID: 22203564.
65. Wang J, Hu B, Xu M, Yan Q, Liu S, Zhu X, Sun Z, Tao D, Ding L, Reed E, Gong J, Li QQ, Hu J. Therapeutic effectiveness of bacteriophages in the rescue of mice with extended spectrum beta-lactamase-producing Escherichia coli bacteremia. Int J Mol Med. 2006;17(2):347-55. PubMed PMID: 16391836; PMCID: 16391836.
66. Regeimbal JM, Jacobs AC, Corey BW, Henry MS, Thompson MG, Pavlicek RL, Quinones J, Hannah RM, Ghebremedhin M, Crane NJ, Zurawski DV, Teneza-Mora NC, Biswas B, Hall ER. Personalized Therapeutic Cocktail of Wild Environmental Phages Rescues Mice from Acinetobacter baumannii Wound Infections. Antimicrob Agents Chemother. 2016;60(10):5806-16. doi: 10.1128/AAC.02877-15. PubMed PMID: 27431214; PMCID: PMC5038255.
67. Rouse MD, Stanbro J, Roman JA, Lipinski MA, Jacobs A, Biswas B, Regeimbal J, Henry M, Stockelman MG, Simons MP. Impact of Frequent Administration of Bacteriophage on Therapeutic Efficacy in an A. baumannii Mouse Wound Infection Model. Front Microbiol. 2020;11:414. doi: 10.3389/fmicb.2020.00414. PubMed PMID: 32256472; PMCID: PMC7090133.
68. Kumari S, Harjai K, Chhibber S. Bacteriophage versus antimicrobial agents for the treatment of murine burn wound infection caused by Klebsiella pneumoniae B5055. J Med Microbiol. 2011;60(Pt 2):205-10. doi: 10.1099/jmm.0.018580-0. PubMed PMID: 20965914.
69. Sarhan WA, Azzazy HM. Apitherapeutics and phage-loaded nanofibers as wound dressings with enhanced wound healing and antibacterial activity. Nanomedicine (Lond). 2017;12(17):2055-67. doi: 10.2217/nnm-2017-0151. PubMed PMID: 28805554.
70. Bull JJ, Otto G, Molineux IJ. In vivo growth rates are poorly correlated with phage therapy success in a mouse infection model. Antimicrob Agents Chemother. 2012;56(2):949-54. doi: 10.1128/AAC.05842-11. PubMed PMID: 22106213; PMCID: PMC3264239.
71. Gurney J, Brown SP, Kaltz O, Hochberg ME. Steering Phages to Combat Bacterial Pathogens. Trends Microbiol. 2020;28(2):85-94. doi: 10.1016/j.tim.2019.10.007. PubMed PMID: 31744662; PMCID: PMC6980653.
72. Vikram A, Woolston J, Sulakvelidze A. Phage Biocontrol Applications in Food Production and Processing. Curr Issues Mol Biol. 2021;40:267-302. doi: 10.21775/cimb.040.267. PubMed PMID: 32644048.
73. Reynaud A, Cloastre L, Bernard J, Laveran H, Ackermann HW, Licois D, Joly B. Characteristics and diffusion in the rabbit of a phage for Escherichia coli 0103. Attempts to use this phage for therapy. Veterinary Microbiology. 1992;30(2-3):203-12. doi: 10.1016/0378-1135(92)90114-9.
74. Jamalludeen N, Johnson RP, Shewen PE, Gyles CL. Evaluation of bacteriophages for prevention and treatment of diarrhea due to experimental enterotoxigenic Escherichia coli O149 infection of pigs. Vet Microbiol. 2009;136(1-2):135-41. doi: 10.1016/j.vetmic.2008.10.021. PubMed PMID: 19058927.
75. Fukuda K, Ishida W, Uchiyama J, Rashel M, Kato S, Morita T, Muraoka A, Sumi T, Matsuzaki S, Daibata M, Fukushima A. Pseudomonas aeruginosa keratitis in mice: effects of topical bacteriophage KPP12 administration. PLoS One. 2012;7(10):e47742. doi: 10.1371/journal.pone.0047742. PubMed PMID: 23082205; PMCID: PMC3474789.
76. Matsuda T, Freeman TA, Hilbert DW, Duff M, Fuortes M, Stapleton PP, Daly JM. Lysis-deficient bacteriophage therapy decreases endotoxin and inflammatory mediator release and improves survival in a murine peritonitis model. Surgery. 2005;137(6):639-46. doi: 10.1016/j.surg.2005.02.012. PubMed PMID: 15933632.
77. Hung CH, Kuo CF, Wang CH, Wu CM, Tsao N. Experimental phage therapy in treating Klebsiella pneumoniae-mediated liver abscesses and bacteremia in mice. Antimicrob Agents Chemother. 2011;55(4):1358-65. doi: 10.1128/AAC.01123-10. PubMed PMID: 21245450; PMCID: PMC3067190.
78. Vinner GK, Rezaie-Yazdi Z, Leppanen M, Stapley AGF, Leaper MC, Malik DJ. Microencapsulation of Salmonella-Specific Bacteriophage Felix O1 Using Spray-Drying in a pH-Responsive Formulation and Direct Compression Tableting of Powders into a Solid Oral Dosage Form. Pharmaceuticals (Basel). 2019;12(1). doi: 10.3390/ph12010043. PubMed PMID: 30909381; PMCID: PMC6469172.
79. Alam M, Akhter MZ, Yasmin M, Ahsan CR, Nessa J. Local bacteriophage isolates showed anti- Escherichia coli O157:H7 potency in an experimental ligated rabbit ileal loop model. Can J Microbiol. 2011;57(5):408-15. doi: 10.1139/W11-020. PubMed PMID: 21542784.
80. Abdulamir AS, Jassim SA, Abu Bakar F. Novel approach of using a cocktail of designed bacteriophages against gut pathogenic E. coli for bacterial load biocontrol. Ann Clin Microbiol Antimicrob.2014;13:39. doi: 10.1186/s12941-014-0039-z. PubMed PMID: 25062829; PMCID: PMC4222638.
81. Tanji Y, Shimada T, Fukudomi H, Miyanaga K, Nakai Y, Unno H. Therapeutic use of phage cocktail for controlling Escherichia coli O157:H7 in gastrointestinal tract of mice. J Biosci Bioeng. 2005;100(3):280-7. doi: 10.1263/jbb.100.280. PubMed PMID: 16243277.
82. Jault P, Leclerc T, Jennes S, Pirnay JP, Que Y-A, Resch G, Rousseau AF, Ravat F, Carsin H, Le Floch R, Schaal JV, Soler C, Fevre C, Arnaud I, Bretaudeau L, Gabard J. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): a randomised, controlled, double-blind phase 1/2 trial. Lancet Infect Dis. 2019;19(1):35-45. doi: 10.1016/s1473-3099(18)30482-1.
83. Sarker SA, McCallin S, Barretto C, Berger B, Pittet AC, Sultana S, Krause L, Huq S, Bibiloni R, Bruttin A, Reuteler G, Brussow H. Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology. 2012;434(2):222-32. doi: 10.1016/j.virol.2012.09.002. PubMed PMID: 23102968.
84. Bruttin A, Brussow H. Human volunteers receiving Escherichia coli phage T4 orally: a safety test of phage therapy. Antimicrob Agents Chemother. 2005;49(7):2874-8. doi: 10.1128/AAC.49.7.2874-2878.2005. PubMed PMID: 15980363; PMCID: PMC1168693.
85. Law N, Logan C, Yung G, Furr CL, Lehman SM, Morales S, Rosas F, Gaidamaka A, Bilinsky I, Grint P, Schooley RT, Aslam S. Successful adjunctive use of bacteriophage therapy for treatment of multidrug-resistant Pseudomonas aeruginosa infection in a cystic fibrosis patient. Infection. 2019;47(4):665-8. doi: 10.1007/s15010-019-01319-0. PubMed PMID: 31102236.
86. Aslam S, Courtwright AM, Koval C, Lehman SM, Morales S, Furr CL, Rosas F, Brownstein MJ, Fackler JR, Sisson BM, Biswas B, Henry M, Luu T, Bivens BN, Hamilton T, Duplessis C, Logan C, Law N, Yung G, Turowski J, Anesi J, Strathdee SA, Schooley RT. Early clinical experience of bacteriophage therapy in 3 lung transplant recipients. Am J Transplant. 2019;19(9):2631-9. doi: 10.1111/ajt.15503. PubMed PMID: 31207123; PMCID: PMC6711787.
87. Maddocks S, Fabijan AP, Ho J, Lin RCY, Ben Zakour NL, Dugan C, Kliman I, Branston S, Morales S, Iredell JR. Bacteriophage Therapy of Ventilator-associated Pneumonia and Empyema Caused by Pseudomonas aeruginosa. Am J Respir Crit Care Med. 2019;200(9):1179-81. doi: 10.1164/rccm.201904-0839LE. PubMed PMID: 31437402.
88. Rose T, Verbeken G, Vos DD, Merabishvili M, Vaneechoutte M, Lavigne R, Jennes S, Zizi M, Pirnay JP. Experimental phage therapy of burn wound infection: difficult first steps. Int J Burns Trauma. 2014;4(2):66-73. PubMed PMID: 25356373; PMCID: PMC4212884.
89. Schooley RT, Biswas B, Gill JJ, Hernandez-Morales A, Lancaster J, Lessor L, Barr JJ, Reed SL, Rohwer F, Benler S, Segall AM, Taplitz R, Smith DM, Kerr K, Kumaraswamy M, Nizet V, Lin L, McCauley MD, Strathdee SA, Benson CA, Pope RK, Leroux BM, Picel AC, Mateczun AJ, Cilwa KE, Regeimbal JM, Estrella LA, Wolfe DM, Henry MS, Quinones J, Salka S, Bishop-Lilly KA, Young R, Hamilton T. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails To Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob Agents Chemother. 2017;61(10). doi: 10.1128/AAC.00954-17. PubMed PMID: 28807909; PMCID: PMC5610518.
90. Ujmajuridze A, Chanishvili N, Goderdzishvili M, Leitner L, Mehnert U, Chkhotua A, Kessler TM, Sybesma W. Adapted Bacteriophages for Treating Urinary Tract Infections. Front Microbiol. 2018;9:1832. doi: 10.3389/fmicb.2018.01832. PubMed PMID: 30131795; PMCID: PMC6090023.
91. Sarker SA, Sultana S, Reuteler G, Moine D, Descombes P, Charton F, Bourdin G, McCallin S, Ngom-Bru C, Neville T, Akter M, Huq S, Qadri F, Talukdar K, Kassam M, Delley M, Loiseau C, Deng Y, El Aidy S, Berger B, Brussow H. Oral Phage Therapy of Acute Bacterial Diarrhea With Two Coliphage Preparations: A Randomized Trial in Children From Bangladesh. EBioMedicine. 2016;4:124-37. doi: 10.1016/j.ebiom.2015.12.023. PubMed PMID: 26981577; PMCID: PMC4776075.
92. Jennes S, Merabishvili M, Soentjens P, Pang KW, Rose T, Keersebilck E, Soete O, Francois PM, Teodorescu S, Verween G, Verbeken G, De Vos D, Pirnay JP. Use of bacteriophages in the treatment of colistin-only-sensitive Pseudomonas aeruginosa septicaemia in a patient with acute kidney injury-a case report. Crit Care. 2017;21(1):129. doi: 10.1186/s13054-017-1709-y. PubMed PMID: 28583189; PMCID: PMC5460490.
93. Aleshkin AV, Ershova ON, Volozhantsev NV, Svetoch EA, Popova AV, Rubalskii EO, Borzilov AI, Aleshkin VA, Afanas’ev SS, Karaulov AV, Galimzyanov KM, Rubalsky OV, Bochkareva SS. Phagebiotics in treatment and prophylaxis of healthcare-associated infections. Bacteriophage. 2016;6(4):e1251379. doi: 10.1080/21597081.2016.1251379. PubMed PMID: 28090384; PMCID: PMC5221749.
94. Wright A, Hawkins CH, Anggard EE, Harper DR. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin Otolaryngol. 2009;34(4):349-57. doi: 10.1111/j.1749-4486.2009.01973.x. PubMed PMID: 19673983.
95. Rhoads DD, Wolcott RD, Kuskowski MA, Wolcott BM, Ward LS, Sulakvelidze A. Bacteriophage therapy of venous leg ulcers in humans: results of a phase I safety trial. J Wound Care. 2009;18(6):237-8, 40-3. doi: 10.12968/jowc.2009.18.6.42801. PubMed PMID: 19661847.
96. Nir-Paz R, Gelman D, Khouri A, Sisson BM, Fackler J, Alkalay-Oren S, Khalifa L, Rimon A, Yerushalmy O, Bader R, Amit S, Coppenhagen-Glazer S, Henry M, Quinones J, Malagon F, Biswas B, Moses AE, Merril G, Schooley RT, Brownstein MJ, Weil YA, Hazan R. Successful Treatment of Antibiotic-resistant, Poly-microbial Bone Infection With Bacteriophages and Antibiotics Combination. Clin Infect Dis. 2019;69(11):2015-8. doi: 10.1093/cid/ciz222. PubMed PMID: 30869755.
97. Pirnay JP, De Vos D, Verbeken G, Merabishvili M, Chanishvili N, Vaneechoutte M, Zizi M, Laire G, Lavigne R, Huys I, Van den Mooter G, Buckling A, Debarbieux L, Pouillot F, Azeredo J, Kutter E, Dublanchet A, Gorski A, Adamia R. The phage therapy paradigm: pret-a-porter or sur-mesure? Pharm Res. 2011;28(4):934-7. doi: 10.1007/s11095-010-0313-5. PubMed PMID: 21063753.
98. Fowler VG, Jr., Das AF, Lipka-Diamond J, Schuch R, Pomerantz R, Jauregui-Peredo L, Bressler A, Evans D, Moran GJ, Rupp ME, Wise R, Corey GR, Zervos M, Douglas PS, Cassino C. Exebacase for patients with Staphylococcus aureus bloodstream infection and endocarditis. J Clin Invest. 2020;130(7):3750-60. doi: 10.1172/JCI136577. PubMed PMID: 32271718; PMCID: PMC7324170.
Submitted May 7, 2022 | Accepted August 9, 2022 | Published October 17, 2022
Copyright © 2022 The Author(s). This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International License.