Delphine Planas1,2, Augustine Fert1,2, Yuwei Zhang1,2, Jean-Philippe Goulet3, Jonathan Richard1,2, Andrés Finzi1,2, Maria Julia Ruiz1,2, Laurence Raymond Marchand2, Debashree Chatterjee1,2, Huicheng Chen1,2, Tomas Raul Wiche Salinas1,2, Annie Gosselin1,2, Eric A. Cohen4, Jean-Pierre Routy5, Nicolas Chomont1,2, Petronela Ancuta1,2,*
1Département de microbiologie, infectiologie et immunologie; Faculté de médecine; Université de Montréal; Montréal, Québec, Canada
2Centre de recherche du CHUM; Montréal, Québec, Canada
3Caprion; Montréal, Québec, Canada
4Institut de recherches cliniques de Montréal; Montréal, Québec, Canada
5Chronic Viral Illness Service; Division of Hematology; McGill University Health Centre-Glen site; Montreal, Québec, Canada
Petronela Ancuta
CHUM-Research Centre
900 rue Saint-Denis, Tour Viger R, Room R09.416
Montreal, Quebec H2X 0A9, Canada;
Phone: 514-890-8000, Extension #35744
e-mail: petronela.ancuta@umontreal.ca
10.20411/pai.v5i1.348
Planas D, Fert A, Zhang Y, Goulet J-P, Goulet J-P, Finzi A, Ruiz MJ, Chatterjee D, Marchand LR, Chen H, Salinas TRW, Gosselin A, Cohen EA, Routy J-P, Chomont N, Ancuta P. Pharmacological Inhibition of PPARy Boosts HIV Reactivation and Th17 Effector Functions, While Preventing Progeny Virion Release and de novo Infection. Pathogens and Immunity. 2020;5(1):177-239. PubMed PMID. doi: 10.20411/pai.v5i1.348
The frequency and functions of Th17-polarized CCR6+RORyt+CD4+ T cells are rapidly compromised upon HIV infection and are not restored with long-term viral suppressive antiretroviral therapy (ART). In line with this, Th17 cells represent selective HIV-1 infection targets mainly at mucosal sites, with long-lived Th17 subsets carrying replication-competent HIV-DNA during ART. Therefore, novel Th17-specific therapeutic interventions are needed as a supplement of ART to reach the goal of HIV remission/cure. Th17 cells express high levels of peroxisome proliferator-activated receptor gamma (PPARy), a transcriptional factor that represses the transcription of the HIV provirus and the rorc gene, which encodes for the Th17-specific master regulator RORyt/RORC2. Thus, we hypothesized that the pharmacological inhibition of PPARy will facilitate HIV reservoir reactivation while enhancing Th17 effector functions. Consistent with this prediction, the PPARy antagonist T0070907 significantly increased HIV transcription (cell-associated HIV-RNA) and RORyt-mediated Th17 effector functions (IL-17A). Unexpectedly, the PPARy antagonism limited HIV outgrowth from cells of ART-treated people living with HIV (PLWH), as well as HIV replication in vitro. Mechanistically, PPARy inhibition in CCR6+CD4+ T cells induced the upregulation of transcripts linked to Th17-polarisation (RORyt, STAT3, BCL6 IL-17A/F, IL-21) and HIV transcription (NCOA1-3, CDK9, HTATIP2). Interestingly, several transcripts involved in HIV-restriction were upregulated (Caveolin-1, TRIM22, TRIM5α, BST2, miR-29), whereas HIV permissiveness transcripts were downregulated (CCR5, furin), consistent with the decrease in HIV outgrowth/replication. Finally, PPARy inhibition increased intracellular HIV-p24 expression and prevented BST-2 downregulation on infected T cells, suggesting that progeny virion release is restricted by BST-2-dependent mechanisms. These results provide a strong rationale for considering PPARy antagonism as a novel strategy for HIV-reservoir purging and restoring Th17-mediated mucosal immunity in ART-treated PLWH.
Keywords: HIV-1; ART; CD4+ T cells; Th17; PPARy; IL-21
The Th17-polarized CD4+ T cells are important players in mucosal immunity and their frequency /function are compromised during HIV infection despite viral-suppressive antiretroviral therapy (ART). Th17 cells are key HIV infection targets and contribute to viral reservoir persistence during ART. This raises the need for novel Th17-specific therapies. In this manuscript, we provide evidence that the pharmacological inhibition of PPARy, a documented repressor of the Th17 master regulator RORyt and HIV transcription, may represent a novel strategy toward Th17-mediated immunity restoration and HIV-reservoir purging in ART-treated PLWH.
Antiretroviral therapies (ART) efficiently control HIV-1 replication to undetectable plasma levels and have improved the life expectancy of people living with HIV (PLWH) [1-3]. However, ART does not cure HIV, with viral rebound occurring rapidly upon treatment interruption [2, 4-6]. In addition, immunological dysregulations persist in ART-treated PLWH leading to an increased risk for non-AIDS co-morbidities such as cardiovascular disease [7] and neurocognitive impairment [8]. Therefore, additional therapeutic interventions to purge viral reservoirs and restore immunological competence in ART-treated PLWH are needed [9].
In ART-treated PLWH, HIV reservoirs persist in a small fraction of long-lived memory CD4+ T- cells [3, 4, 10-12] and likely other cellular/anatomic reservoirs [13]. Studies by our group and others demonstrated that among CD4+ T cells, Th17-polarized cells are strategically located at portal sites of HIV/SIV entry and efficiently support integrative HIV infection [14-16]. Moreover, Th17 cells are depleted from the gut-associated lymphoid tissues during HIV/SIV infection, and their frequency is not restored with ART [14, 15]. This causes dramatic alterations in mucosal barrier integrity, increased microbial translocation from the gut, and systemic immune activation [14, 15], all leading to non-AIDS co-morbidities [7, 8]. Although the depletion of mucosal Th17 cells is well-documented during HIV/SIV infection, a fraction of Th17 cells is long lived and enriched in HIV-DNA in the blood and colon of ART-treated PLWH [14, 15]. The key role played by Th17 cells in mucosal homeostasis, their contribution to HIV persistence, as well as the deleterious consequences of their paucity in ART-treated PLWH, indicate that the design of novel Th17-specific therapeutic strategies is needed for HIV remission/cure [14, 15].
Th17 cells are distinguished from the other CD4+ T-cell subsets by a unique transcriptional signature that includes multiple HIV permissiveness factors (eg, CCR5, NF-kB, mTOR, NFATC2IP), the lack of anti-HIV defense mechanisms [14, 15], as well as the peroxisome proliferator-activated receptor gamma (PPARy) [17-20]. PPARy is an intrinsic negative regulator of NF-kB [21] and an inhibitor of HIV transcription [17, 22-24]. PPARy is a member of the PPAR subfamily of ligand-dependent non-steroid nuclear receptors; PPARy forms an obligatory heterodimer with retinoic X receptor (RXR) and binds onto PPAR responsive elements (PPREs) expressed on the promoters /regulatory regions of specific genes, thus functioning as a transcriptional repressor or activator [25, 26]. PPARy is expressed by multiple immune and non-immune cells and acts as a lipid sensor that controls the expression of numerous genes involved in lipid/glucose metabolism. Natural and synthetic PPARy agonists have been documented to regulate metabolic/inflammatory processes [26-29], in part via the mTOR activation pathway [30]. It is noteworthy that PPREs are present in the HIV long terminal repeat (LTR) region, indicating that PPARy participates directly in the negative regulation of HIV transcription [31]. Increasing evidence supports a role of PPARy in the regulation of adaptive immunity by acting on T-cell proliferation and differentiation [27, 29, 32-34]. Of particular importance, it was reported that PPARy inhibits Th17 effector functions by the transcriptional repression of RORyt [32, 34], the master regulator of Th17 differentiation [14, 15].
Clinical trials were previously performed using PPARy agonists/activators, for example, rosiglitazone (RGZ) for treating the lypodystrophy caused by specific classes of antiretroviral drugs [35], as well as metabolic syndrome and inflammation in HIV-infected individuals [36-39]. However, to our knowledge, no clinical trials were performed using PPARy targeting drugs in the context of HIV cure/remission strategies. Although the PPARy activation blocks HIV replication in primary T cells [17], with PPARy agonists being expected to promote deep latency, studies in SIV-infected rhesus macaques demonstrated that hematopoietic alterations caused by Nef are dependent on the PPARy activation and are mimicked by the PPARy agonist RGZ [40]. Based on this evidence, Prost et al. proposed that PPARy inhibition may be more appropriate to counteract hematopoietic alterations caused by HIV/SIV infections [40] and emphasized the need for the development of clinically advanced PPARy antagonists [41]. Of particular importance, the pharmacological inhibition of PPARy may promote HIV reservoir reactivation, in a manner similar to that of currently tested latency reversing agents (LRA) [42, 43]. This scenario is supported by our previous studies demonstrating that RNA interference against PPARy increases permissiveness to viral replication in vitro [17].
In this study, we investigated the effect of PPARy pharmacological inhibition on HIV reservoir reactivation and immune function restoration in Th17 cells, a subset enriched in PPARy mRNA and protein [17, 18]. Our results demonstrate that the PPARy antagonism increased both HIV transcription and RORyt-mediated Th17 effector functions, such as IL-17A and IL-21 production", in CD4+ T cells from ART-treated PLWH. Of note, IL-21 is a signature-cytokine for follicular helper T-cells (Tfh) [33] that is also key for Th17 survival [14] and has demonstrated antiviral activity in vitro [44] and in non-human primate models [45, 46]. Unexpectedly, the PPARy antagonism limited viral outgrowth in CD4+ T cells of ART-treated PLWH ex vivo, as well as on HIV infection in vitro. Thus, unique combination of these immunological and virological features provides a strong rationale for considering the pharmacological inhibition of PPARy for HIV cure/remission strategies.
PLWH receiving viral-suppressive ART (Table 1) and HIV- individuals (n=13 males; n=2 females) were recruited at the Montreal Chest Institute, McGill University Health Centre and Centre Hospitalier de l’Université de Montréal (CHUM) in Montreal, Quebec, Canada. Large quantities of PBMCs (109–1010 cells) were collected by leukapheresis, as previously described [19, 20].
Table 1: Clinical parameters of ART-treated PLWH study participants.
|
Patient |
Sex |
CD4 count# |
CD8 count# |
Plasma viral load& |
Time since infection* |
ART |
Time on ART* |
|
ART #1 |
M |
398 |
775 |
<40 |
154 |
Complera |
<26 |
|
ART #2 |
M |
841 |
1,322 |
<40 |
150 |
Sustiva/Truvada |
138 |
|
ART #3 |
M |
796 |
399 |
<40 |
8 |
Stribild |
6 |
|
ART #4 |
M |
581 |
1,060 |
<40 |
96 |
Sustiva/Truvada |
5 |
|
ART #5 |
M |
391 |
620 |
50 |
165 |
Kivexa/Delavirdine |
54 |
|
ART #6 |
M |
318 |
431 |
<40 |
149 |
Kivexa/Delavirdine |
44 |
|
ART #7 |
M |
514 |
568 |
<40 |
16 |
Tivicay/ Truvada |
6 |
|
ART #8 |
M |
775 |
1,000 |
<40 |
74 |
Complera |
19 |
|
ART #9 |
M |
459 |
545 |
<40 |
189 |
Truvada/Raltegravir |
>12 |
|
ART #10 |
F |
616 |
330 |
<40 |
186 |
Viracept/Truvada |
34 |
|
ART #11 |
M |
542 |
803 |
<40 |
13 |
Stribild |
12 |
|
ART #12 |
M |
458 |
899 |
<40 |
201 |
Truvada/Viramune |
200 |
|
ART #13 |
M |
908 |
854 |
<40 |
89 |
Stribild |
72 |
|
ART #14 |
F |
833 |
445 |
<40 |
213 |
Viracept/Truvada |
60 |
|
ART #15 |
M |
546 |
1,116 |
<40 |
408 |
Atripla |
372 |
#, cells/µl; &, HIV-RNA copies per ml plasma; *, months; ART, antiretroviral therapy; M, male; F, female
This study, using PBMCs from HIV-uninfected and HIV-infected study participants was conducted in compliance with the principles included in the Declaration of Helsinki. This study received approval from the Institutional Review Board (IRB) of the McGill University Health Centre and the IRB of the CHUM-Research Centre, Montreal, Quebec, Canada. All participants signed a written informed consent and agreed with the publication of the results generated using their biological samples.
The following drugs were used: T0070907 (T007; 2-Chloro-5-nitro-N-4-pyridinylbenzamide; Tocris, Cayman Chemical, Michigan, USA); rosiglitazone (RGZ; Cayman Chemical, Michigan, USA); Saquinavir, and Raltegravir (NIH AIDS Reagent Program, Maryland, USA).
The fluorochrome-conjugated antibodies used for polychromatic flow cytometry are listed in Supplementary Table 3. A viability dye (Molecular Probes® LIVE/DEAD® Fixable Dead Cell Stain Kits, Invitrogen) was used to exclude dead cells. Intracellular staining was performed using Fixation/Permeabilization Solution Kit (BD). Cells were analyzed using an LSRII cytometer, Diva version 6 (BD Biosciences, San Jose, CA), and FlowJo version 10.0.6 (Tree Star, Inc). Flow cytometry gates were defined using the fluorescence minus one (FMO) strategy [19, 20].
Total and memory CD4+ T cells were enriched from PBMCs by negative selection using magnetic beads (magnetic-activated cell sorting [MACS], Miltenyi), with a purity of >95%, as previously described [19, 20]. Highly pure CCR6+/CCR6- T cells were sorted by FACS using the antibodies listed in Supplementary Table 3, as previously reported by our group [19, 20].
A viral outgrowth assay (VOA) was performed using a protocol previously established by our group [19, 20]. Briefly, total memory CD4+ T cells isolated by MACS from PBMCs of PLWH receiving viral-suppressive ART (PLWH+ART) were cultured (RPMI1640, 10% FBS, 1% antibiotics) at 1×106 cells/mL/well in 48-well plates in the presence of immobilized CD3 and soluble CD28 antibodies (1 µg/mL) for up to 12 days. At day 3, cells were washed, split into 2 new wells, and cultured with IL-2 (5 ng/mL). At days 6 and 9, cells from each well were split into 2 new wells, and media was refreshed. Supernatants were collected at days 3, 6, 9, and 12 for HIV-p24 and cytokine quantification by ELISA. At day 12, cells were stimulated with PMA (50 ng/mL) and Ionomycin (1ug/mL) in the presence of Brefeldin A (5 µg/mL) for 5 hours and used for the intracellular detection of HIV-p24, IL-17A, and IFN-y by flow cytometry after staining with specific antibodies (Supplementary Table 3).
Cell-associated (CA) RNA and DNA were dually extracted from cell pellets (5-6 replicates of 1×106 cells/experimental condition were pooled) using the AllPrep DNA/RNA Mini Kit (Qiagen), according to the manufacturer’s instructions. The quality (260 nm/280 nm ratio) and quantity of RNA/DNA collected were evaluated by Nanodrop.
CA LTR-Gag HIV-RNA (CA HIV-RNA) levels were quantified by 1-step real-time RT-PCR using specific external/internal primers and taqman probes (Supplementary Table 4A) and classical RT-PCR/PCR amplification conditions. The amplified products from the first PCR (ProFlex PCR System 9700; Applied Biosystems) were diluted 1:10 x in molecular grade water and used as templates in second nested real-time PCR amplifications (RotorGene instrument, Qiagen). For the CA LTR-Gag HIV-RNA (unspliced), standards were generated using plasmid-based transcription in vitro (MEGAscript™ T7 Transcription Kit, ThermoFisher).
To normalize HIV-RNA to HIV-DNA on matched samples, levels of CA Gag HIV-DNA were quantified by ultrasensitive nested real-time PCR using the same primers and Taqman probe used for the CA HIV-RNA quantification (Supplementary Table 4A). To normalize the HIV-DNA levels per number of cells, the CD3 gene was concomitantly amplified using specific external/internal primers and Taqman probes (Supplementary Table 4B), as previously described [19, 20]. ACH2 cells carrying 1 copy of integrated HIV-DNA per cell (The National Institutes of Health AIDS Reagent Program) were used for the standard curve.
The quantification of cell-free (CF) HIV-RNA was performed as previously reported (48). To concentrate HIV virions, 5 mL aliquots of cell culture supernatants were centrifuged at 25,000g for 90 minutes. Pelleted virions (in 140 µL supernatant) were used for total RNA isolation using the QIAamp Viral RNA Mini Kit (Qiagen; final elution in 60 µL). The extracted RNA was first subjected to DNase (Invitrogen) treatment. HIV-RNA quantification was performed as described above. HIV-RNA quantification was performed in triplicate (using 17 µL eluted total RNA/test), as described above. Results are expressed as the number of HIV-RNA copies per reaction (equivalent of 5 mL cell culture supernatant per test). Standards were generated using RNA extracted from ACH2 culture supernatant. All measures were performed in triplicate.
T cells were activated with CD3/CD28 antibodies (1 µg/mL), exposed to the replication-competent transmitted/founder (T/F) strain HIV THRO (NIH AIDS Reagent Program) [93], and viral replication monitored by ELISA, as previously described [19, 20]. Infected cells were cultured with IL-2 (5 ng/mL) in the presence or absence of T0070907 (1 µM, 5 µM, 10 µM) or RGZ (50 µM). In parallel, experiments were performed with single round VSV-G-pseudotyped HIV (VSV-G/HIV; NL4.3 backbone, env-, gfp in place of nef) [57]. The viral stocks were produced by transfection of 293T cells, as previously described [17, 18].
Integrated HIV-DNA was quantified by ultrasensitive nested real-time PCR in cell lysates (105 cells/test in triplicate; detection limit: 3 HIV-DNA copies/test), with normalization relative to CD3 copy numbers (2 CD3 copies per single cell), as previously described [12, 19, 20], using specific primers and FRET probes (Supplementary Tables 3C-D).
Total RNA was isolated using the RNeasy Kit (Qiagen) and quantified using the Pearl nanophotometer (Implen). One step SYBR Green real-time RT-PCR (Qiagen) was carried out in a LightCycler 480 II (Roche) according to the manufacturer’s recommendations, as we previously reported [17, 18]. QuantiTect Primer Assays were purchased from Qiagen. The expression of each gene was normalized relative to 28S rRNA levels. Amplifications were performed in triplicate on 70 ng RNA/test for target genes and 2 ng RNA/test for 28S rRNA.
Genome-wide transcriptional profiling was performed on total RNA by Genome Québec (Montréal, Québec, Canada) using the Illumina RNA-Sequencing model HiSeq 4000 PE100. Briefly, the paired-end sequencing reads were aligned to coding and non-coding transcripts from Homo Sapiens database GRCh 37 version75 and quantified using the kallisto software version 0.44.0 [94]. The entire RNA-Sequencing data set and the technical information requested by Minimum Information About a Microarray Experiment (MIAME) are available at the Gene Expression Omnibus database under accession GSE128121. One-way ANOVA analysis identified differentially expressed genes based on P values (P<0.05) or adjusted P values (adj. P<0.05) and/or fold-change (FC, cutoff 1.3). Statistical analyses were performed using R version 3.5.1. Differential expression analysis was performed using the limma Bioconductor package [95] (version 3.38.3) on the log2-counts per million (logCPM) transformed transcript-level data. Gene set enrichment analysis was performed using the GSVA method [96] (package version 1.30.0) on the logCPM data using a Gaussian cumulative distribution function.
All statistical analyses were performed using the Prism 8 (GraphPad software). Specifications on the statistical test used are included on the graphs and Figure legends. P values are indicated on the graphs with statistical significance as follows: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
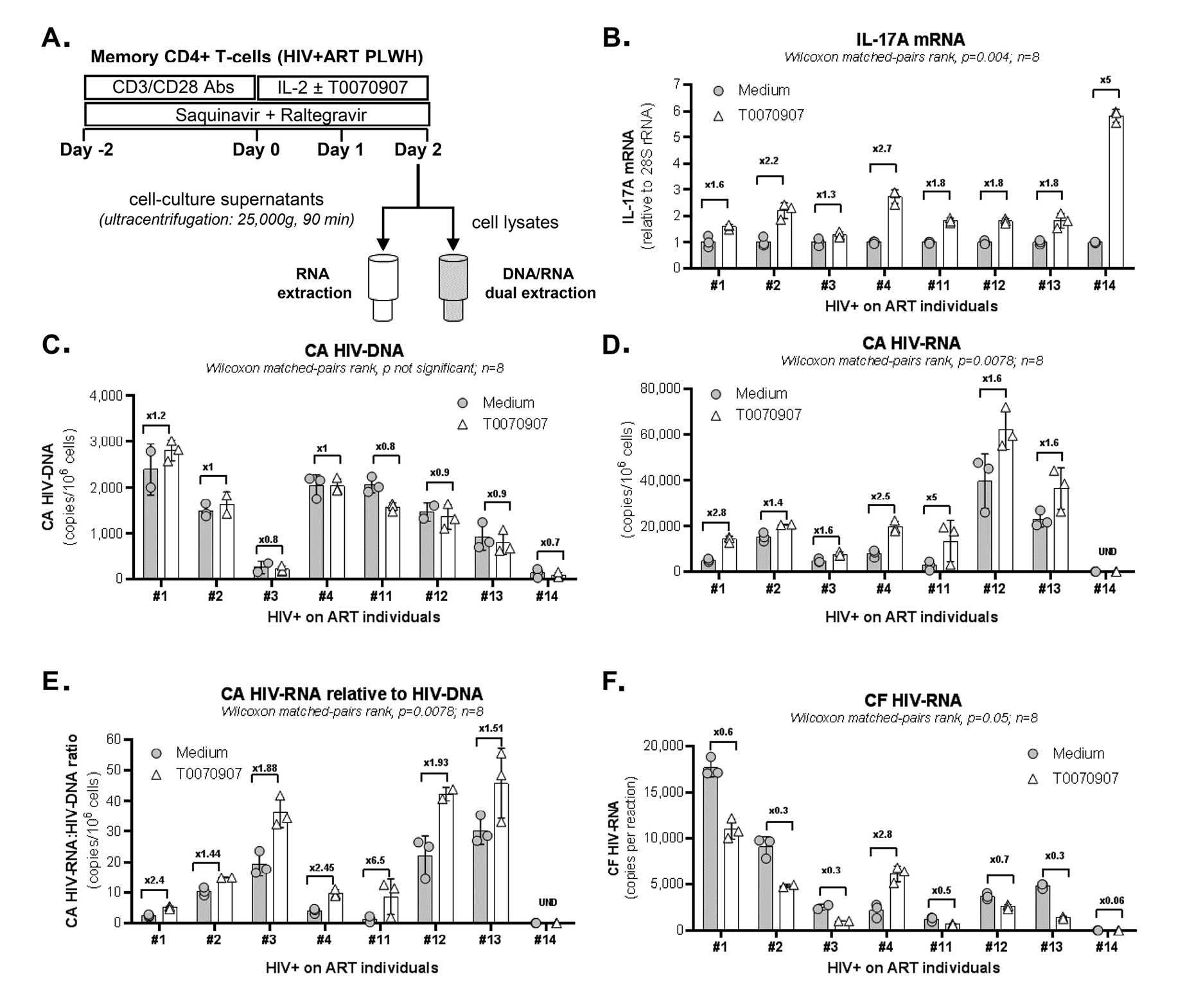
We hypothesized that PPARy pharmacological inhibition promotes both HIV reservoir reactivation and immune function restoration in Th17 cells. To test this hypothesis, we explored the effects of the well-characterized PPARy antagonist T0070907 [47] in memory CD4+ T cells from ART-treated PLWH (Table 1, n=8) (Figure 1A). Cells were stimulated with CD3/CD28 antibodies for 2 days to induce HIV optimal outgrowth [48] and PPARy expression (Supplementary Figure 1) [17]; cells were further cultured in the presence/absence of T0070907 for 2 additional days. To study the post-integration steps of viral replication (ie, transcription, virion production and release) while preventing novel infection in vitro, experiments were performed in the presence of the antiretroviral drugs (ARV) Saquinavir and Raltegravir (Figure 1A). Preliminary experiments allowed the identification of an optimal T0070907 concentration (ie, 10 µM) that upregulates IL-17A production without affecting cell viability and proliferation (Supplementary Figure 2A). As expected, exposure to T0070907 resulted in a significant increase of IL-17A mRNA levels (Figure 1B). During this short-term stimulation/culture in vitro, CA HIV-DNA levels remained similar in T cells cultured with or without T0070907 (Figure 1C), consistent with the well-established stability of HIV-DNA reservoirs [4, 10]. Nevertheless, exposure to T0070907 significantly increased absolute CA HIV-RNA levels, as well as CA HIV-RNA:HIV-DNA ratios (Figure 1D-E), indicating that the drug boosted the TCR-mediated HIV transcription. Unexpectedly, CF HIV-RNA levels were significantly reduced by T0070907 in 7 of 8 donors (Figure 1F), indicative of a post-transcriptional block in virion production and/or release. Thus, the PPARy antagonism overcomes the PPARy-mediated repression of RORyt and HIV transcription, but also modulates the expression of yet unidentified factors that likely act at the post transcriptional level, hence resulting in decreased de novo production and release of viral particles.
Figure 1. The PPARy antagonist T0070907 increases HIV and IL-17A transcription, but inhibits viral release from memory CD4+ T cells of ART-treated PLWH. (A) Shown is the experimental flow chart. Briefly, memory CD4+ T cells of ART-treated PLWH (Table 1, n=8) were activated by CD3/CD28 for 2 days in the presence of ARV (Saquinavir 5 µM; Raltegravir 200 µM) to limit cell-to-cell virion spreading, washed and further cultured with ARV in the presence or the absence of T0070907 (10µM) for anther 48 hours. DMSO (1 µL/µL; identified as Medium) was used as a control. Total RNA and DNA levels were dually extracted from cell pellets and total RNA was extracted from cell culture supernatants. (B) IL-17A mRNA was quantified by real-time RT-PCR and normalized to 28S rRNA levels. (C) Cell-associated (CA) HIV-DNA (Gag primers) was quantified by nested real-time PCR and normalized per 106 cells (2 copies of CD3-DNA per cell). (D-E) CA HIV-RNA (unspliced, Gag primers) levels were quantified by nested real-time RT-PCR and normalized per 106 cells (D) and HIV-DNA/106 cells (E) using results from panel C. (F) Cell-free (CF) HIV-RNA (Gag primers) copies were quantified by nested real-time RT-PCR in RNA extracted from cell culture supernatants. Each symbol represents 1 experimental replicate (mean±SD). The Wilcoxon matched-pairs signed rank test P-values and the FC ratios between medium and T0070907 are indicated on the graphs.
Productive HIV replication is regulated at multiple post-transcriptional steps [1]. To further document the effect of PPARy antagonism on de novo HIV production, a VOA that monitors viral reservoir reactivation and cell-to-cell propagation in culture [19, 20] was performed (Figure 2A). To optimally detect replication-competent HIV, memory CD4+ T cells were isolated from PLWH receiving ART for for more than two years (#5, #10, #12, and #15) and for less than two years (#3 and #4) (Table 1). In a first set of experiments, HIV outgrowth was measured by intracellular HIV-p24 staining at day 12 post-stimulation in cells from 8 splitting replicates merged together (generated from 1 original replicate). Results in Figure 2B-C demonstrate that the HIV outgrowth induced by CD3/CD28 triggering was significantly reduced in the presence of T0070907, with no significant impact on cell viability (Figure 2D). By merging the cells from the 8 identical replicates, it was possible to stimulate the cells with PMA/Ionomycin and monitor the expression of HIV-p24 in cells producing IL-17A and/or IFN-y. Consistent with the well-documented Th17 cell permissiveness to HIV [14, 15], when the VOA was performed in the absence of T0070907, the highest frequency of infected cells was detected in Th17 (IL-17A+IFN-y-) and Th1Th17 (IL-17A+IFN-y+) cells; T0070907 reduced the frequency of HIV-p24+, but not IL-17A+ cells (data not shown). These results indicate the ability of T0070907 to limit HIV replication in Th17 cells without altering their effector functions.
Considering the stochastic distribution of HIV reservoirs, the VOA was performed again with cells from n=4 ART-treated PLWH (Table 1; ART #3, #4, #12, and #15), but this time using 4 original replicates of 106 cells/well (Figure 2E-F) instead of 1 (Figure 2B-D). The HIV-p24 ELISA quantification was performed in cell culture supernatants collected at days 3, 6, 9, and 12 post-stimulation from all splitting replicates. Results in Figure 2E-F confirm the capacity of T0070907 to inhibit HIV outgrowth.
Given the documented ability of RGZ in inhibiting HIV replication [17, 49] by repressing HIV transcription [31], we used RGZ as a control in this VOA. As expected, RGZ (50 µM, optimal dose previously identified [17]) inhibited viral outgrowth in cells of ART-treated PLWH (Table 1; ART #3, #4, #5, and #10) (Supplementary Figure 3A), with no significant effects on cell viability (Supplementary Figure 3B).
Thus, the PPARy antagonism inhibits viral outgrowth by acting on viral replication steps downstream of transcription, steps that are important for de novo viral particle production and/or propagation and spread.
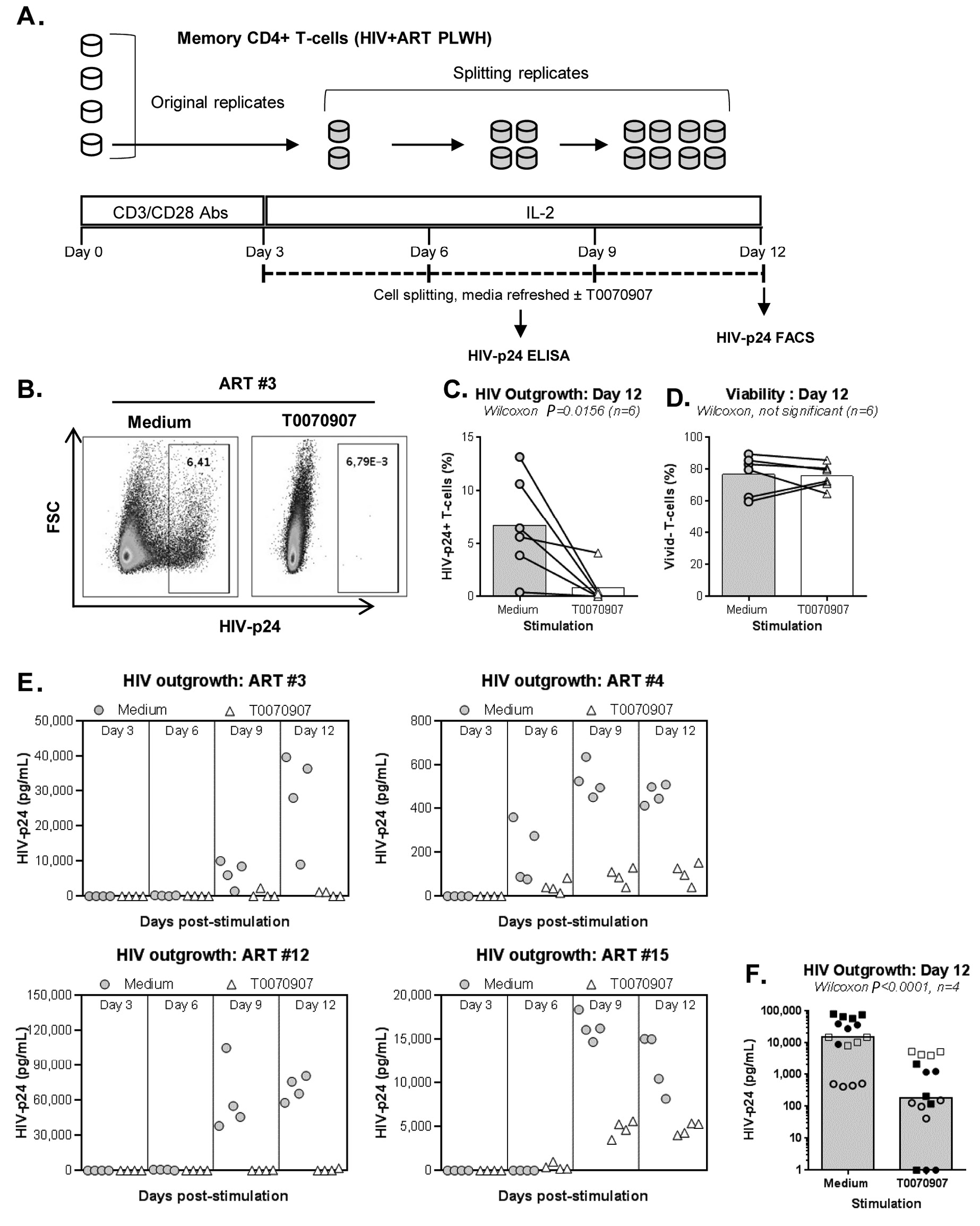
Figure 2. T0070907 inhibits HIV outgrowth in memory CD4+ T cells of ART-treated PLWH. (A) Shown is the experimental flow chart for the viral outgrowth assay (VOA) performed with memory CD4+ T cells of ART-treated PLWH. Briefly, cells cultured in 48-well plates (106 cells/well) were activated with CD3/CD28 antibodies for 3 days, washed and cultured in the presence or the absence of T0070907 (10 µM) up to 12 days. Cells were split into 2 new wells, supernatants collected and media refreshed every 3 days. At day 12, cells were stained with a viability dye and then intracellularly with HIV-p24 antibodies. (B-D) In a first set of experiments, the VOA was performed with one original replicate (106 cells/well) at day 0 that generated 8 splitting replicates at day 12. Shown is (B) the intracellular HIV-p24 expression in cells pooled from the 8 splitting replicates at day 12 from one representative donor (ART #3), as well as statistical analysis of (C) intracellular HIV-p24 staining and (D) cell viability in n=6 ART-treated PLWH (Table 1; ART #3, #4, #5, #10, #12, and #15) (E-F). In another set of experiments, the VOA was performed in 4 original replicates of 106 cells/well cultured at day 0 that each generated 8 splitting replicates at day 12. Shown are HIV-p24 levels in cell culture supernatant quantified in cell culture supernatant collected from the splitting replicates of each original replicate at days 3 (1 well), 6 (2 wells), 9 (4 wells), and 12 (8 wells) for each donor individually (E) and statistical analysis on n=4 ART-treated PLWH at day 12 (F) (Table 1; ART #3, #4, #12, and #15). Each symbol represents the median HIV-p24 value of 8 splitting replicate wells resulting from 1 original replicate (4 original replicates/donor), with grey circles for Medium and open triangles for T0070907 (E) and different symbols for each donor (F). The Wilcoxon matched-pairs signed rank test P-values and the fold change (FC) ratios between medium and T0070907 are indicated on the graphs.
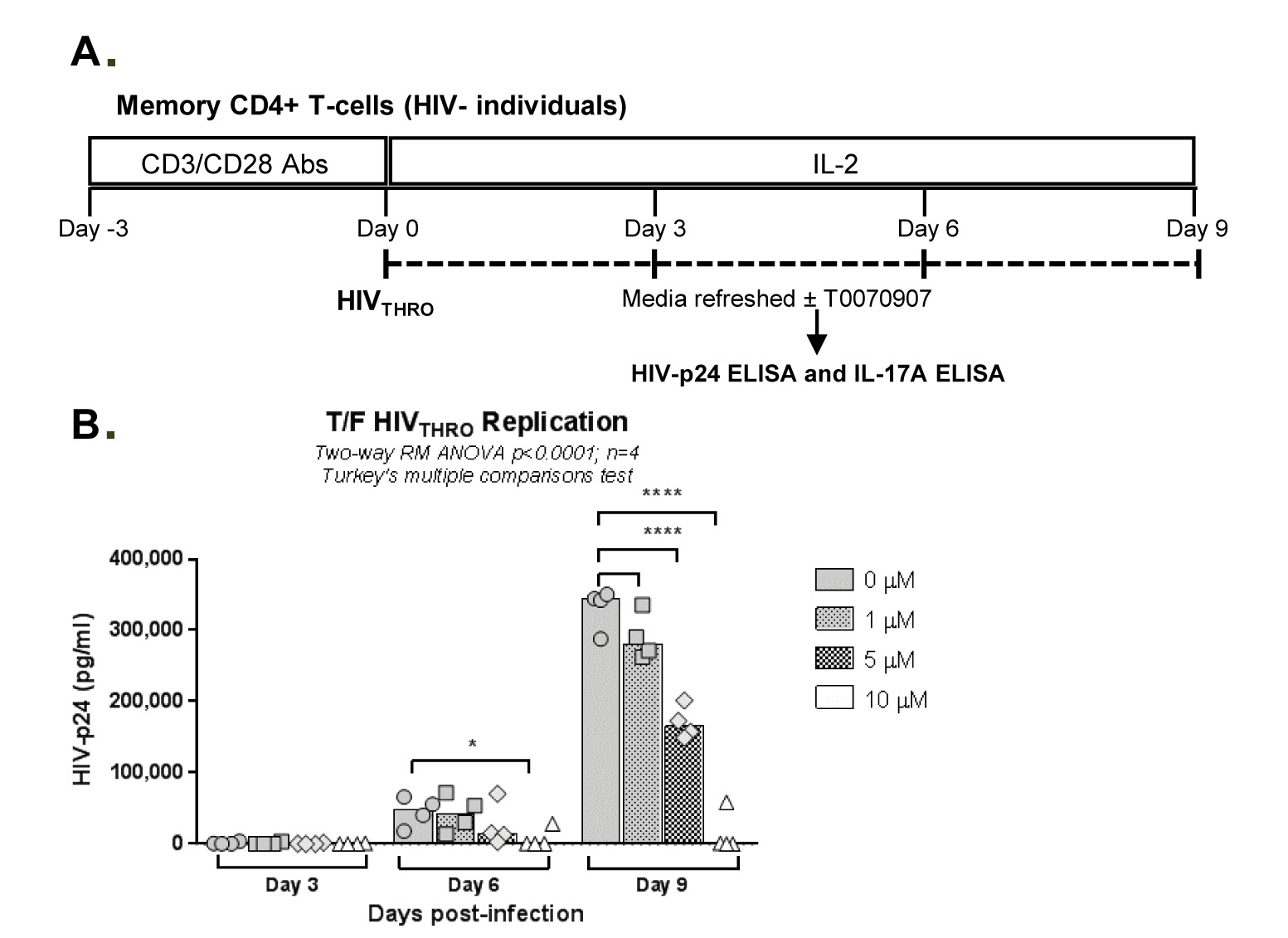
Figure 3. T0070907 boosts IL-17A production and limits T/F HIVTHRO replication in vitro in a dose-dependent manner. (A) Shown is the experimental flow chart. Briefly, memory CD4+ T cells isolated from HIV-uninfected individuals were stimulated by CD3/CD28 for 3 days. (B-C) Cells were exposed to T/F HIVTHRO strain (25 ng/106 cells) and cultured in the presence of IL-2 (5 ng/mL) and in the presence/absence of T0070907 (1, 5, and 10 µM) for up to 9 days, with media, IL-2 in the presence or the absence of T0070907 being refreshed every 3 days. Shown are HIV-p24 levels (B) and IL-17A (C) quantified by ELISA in cell culture supernatants at days 3, 6 and 9 post-infection (n=4). Each symbol represents 1 different donor, and bars represent median values. Two-way RM ANOVA P-values and Turkey’s multiple comparisons are indicated on the graphs. (D) To determine the effect of single versus multiple T0070907 doses on HIV replication, in another set of experiments, infected cells were cultured in the presence of IL-2 and in the presence/absence of T0070907 (5 and 10 µM), with T0070907 being administered either once at day 0 post-infection (0), twice at days 0 and 6 post-infection (0-6), or every 3 days post-infection (0-3-6). Shown are relative HIV-p24 levels quantified by ELISA in cell culture supernatants collected at day 9 post-infection (n=3).
Considering the unexpected antiviral features of T0070907, we further investigated its ability to modulate HIV replication in vitro. For this, we used the transmitted/founder (T/F) strain THRO, documented to exhibit high virulence [50], using the experimental design depicted in Figure 3A. TCR-activated memory CD4+ T cells were infected with HIVTHRO and treated with T0070907 (1, 5, 10 µM) for up to 9 days, with T0070907 being refreshed in the media every 3 days. Results indicate a dose-dependent effect of T0070907, with a significant increase in IL-17A production and a decrease in HIV replication observed at 10 µM (Figure 3B-C), in the absence of effects on cell viability and proliferation (Supplementary Figure 2A). In parallel, similar experiments were performed with T0070907 being added every 3 days versus once (day 0 post-infection) or twice (days 0 and 6 post-infection). Results in Figure 3D clearly demonstrate that the antiviral effect of T0070907 is achieved with a single dose of T0070907 added immediately on infection. No effects on cell viability and proliferation were observed (Supplementary Figure 2B). This is indicative that PPARy inhibition during the early steps of infection allows a robust control of HIV spread in culture.
To get insights into the mechanisms of T0070907 action, we investigated its effect on the expression of the HIV receptor CD4 and co-receptors CCR5/CXCR4. Although T0070907 did not change CD4 and CXCR4 expression, a significant decrease in CCR5 expression was observed (Supplementary Figure 4A-D). Thus, in addition to reducing viral production/release (Figure 1F), T0070907 also limits de novo infection in part by limiting CCR5-mediated HIV entry.
IL-17A production and HIV permissiveness are key features of memory CCR6+CD4+ T-cells (14-16). Thus, we further tested the immunological/virological effects of T0070907 in flow cytometry-sorted memory CCR6+ and CCR6- T cells on HIV infection in vitro (Figure 4A). In the absence of T0070907, CCR6+ versus CCR6- T cells expressed significantly higher levels of IL-17A and CCR5 mRNA (Figure 4B-C, left panels) and supported a more robust HIV-DNA integration (≈2 log10 difference) (Figure 4D, left panel). Similar to results on bulk memory T cells, T0070907 significantly increased IL-17A mRNA expression (Figure 4B, right panel) and reduced CCR5 mRNA expression as well as HIV-DNA integration in memory CCR6+ T cells (Figure 4C-D, right panels). Thus, consistent with superior expression of PPARy in CCR6+ Th17/Th1Th17-polarized versus CCR6- Th1-polarized T cells [17, 18, 32, 34], T0070907 acted on CCR6+ T cells to upregulate IL-17A production and limit HIV de novo infection by mechanisms including CCR5 downregulation.
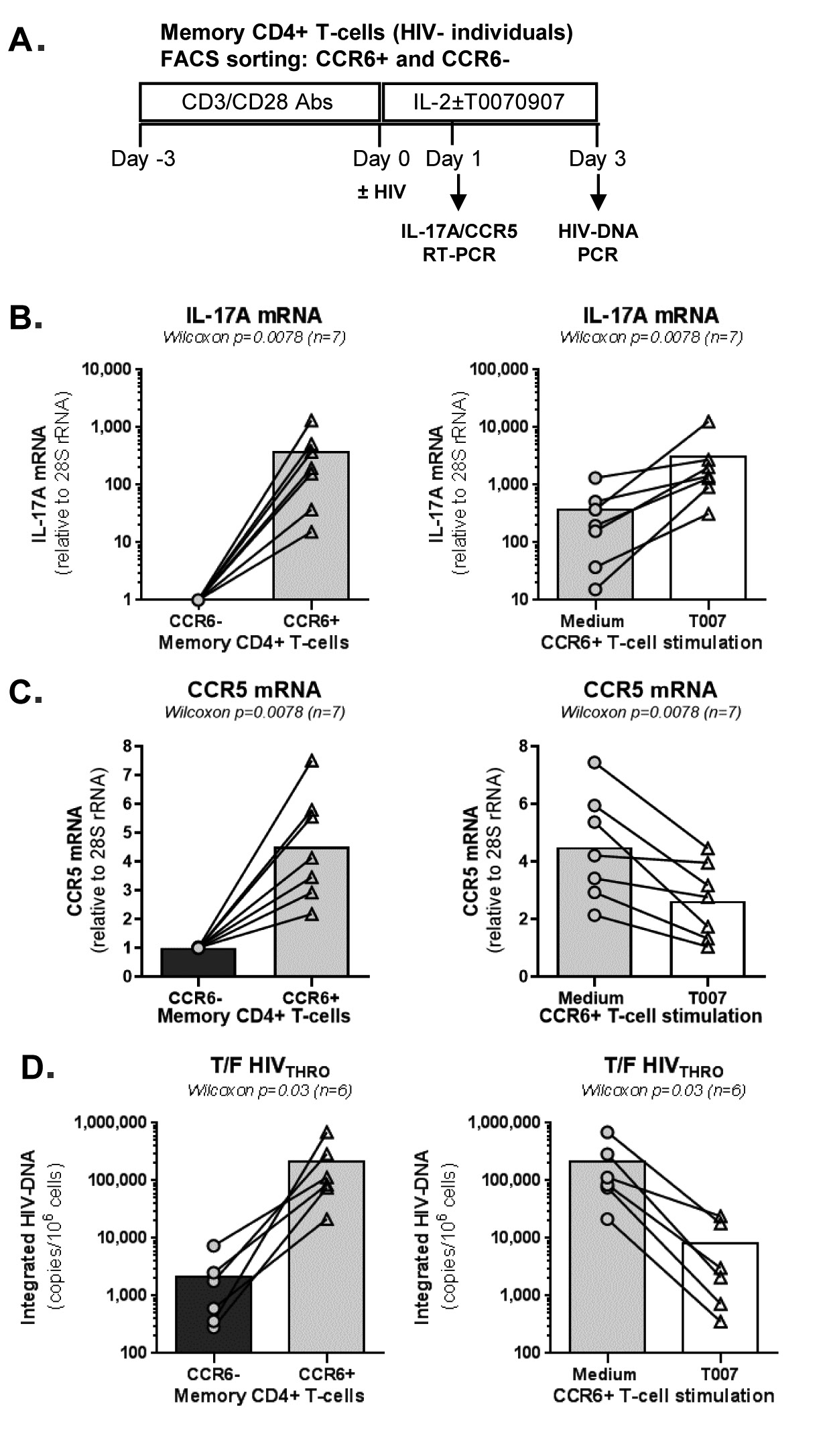
Figure 4. T0070907 efficiently increases IL-17A expression and reduces HIV replication in sorted memory CCR6+ T cells. (A) Shown is the experimental flow chart. Briefly, memory CCR6+ and CCR6- T cells of HIV-uninfected individuals (n=6-7) were stimulated by CD3/CD28 for 3 days. (B-C) Cells were cultured in the presence of IL-2 of T0070907 (10 mM) for 18 hours and RNA extraction was performed for RT-PCR quantification. Shown are results on (B) IL-17A (n=7) and (C) CCR5 (n=7) mRNA expression in CCR6- versus CCR6- T cells cultured in the absence of T0070907 (left panels) and CCR6+ T cells cultured in the presence/absence of T0070907 (right panels). Normalization was performed relative to 28S rRNA, with expression in CCR6- T cells being considered 1. (D) Another fraction of cells was exposed to T/F HIVTHRO strain (25 ng/106 cells) and cultured in the presence of IL-2 of T0070907 (10 mM) for 3 additional days. Shown are levels of HIV-DNA integration (as a measure of HIV replication) in CCR6- versus CCR6+ T cells in the absence of T0070907 (left panel) and in CCR6+ T cells cultured in the presence/absence of T0070907 (right panel). The Wilcoxon signed rank test P-values are indicated on the graphs. Each symbol represents results generated with cells from one different donor; bars represent median values.
To get further insights into the mechanism of action of PPARy antagonism, genome-wide transcriptional profiling was performed in CCR6+ T cells stimulated via the TCR for 3 days and cultured in the presence or absence of T0070907 for an additional 18 hours (Figure 5A). Differentially expressed genes were classified based on P or adjusted P and FC gene expression. Profound transcriptional changes were induced by T0070907 in CCR6+ T cells, with 4,002 transcripts upregulated and 1,249 transcripts downregulated (adj. P<0.05; FC cutoff, 1.3) (Figure 5B), and the top 50 upregulated (adj. P<0.05; FC>8) and downregulated (adj. P<0.05; FC<-3.2) transcripts listed in Supplementary Tables 1-2, respectively.
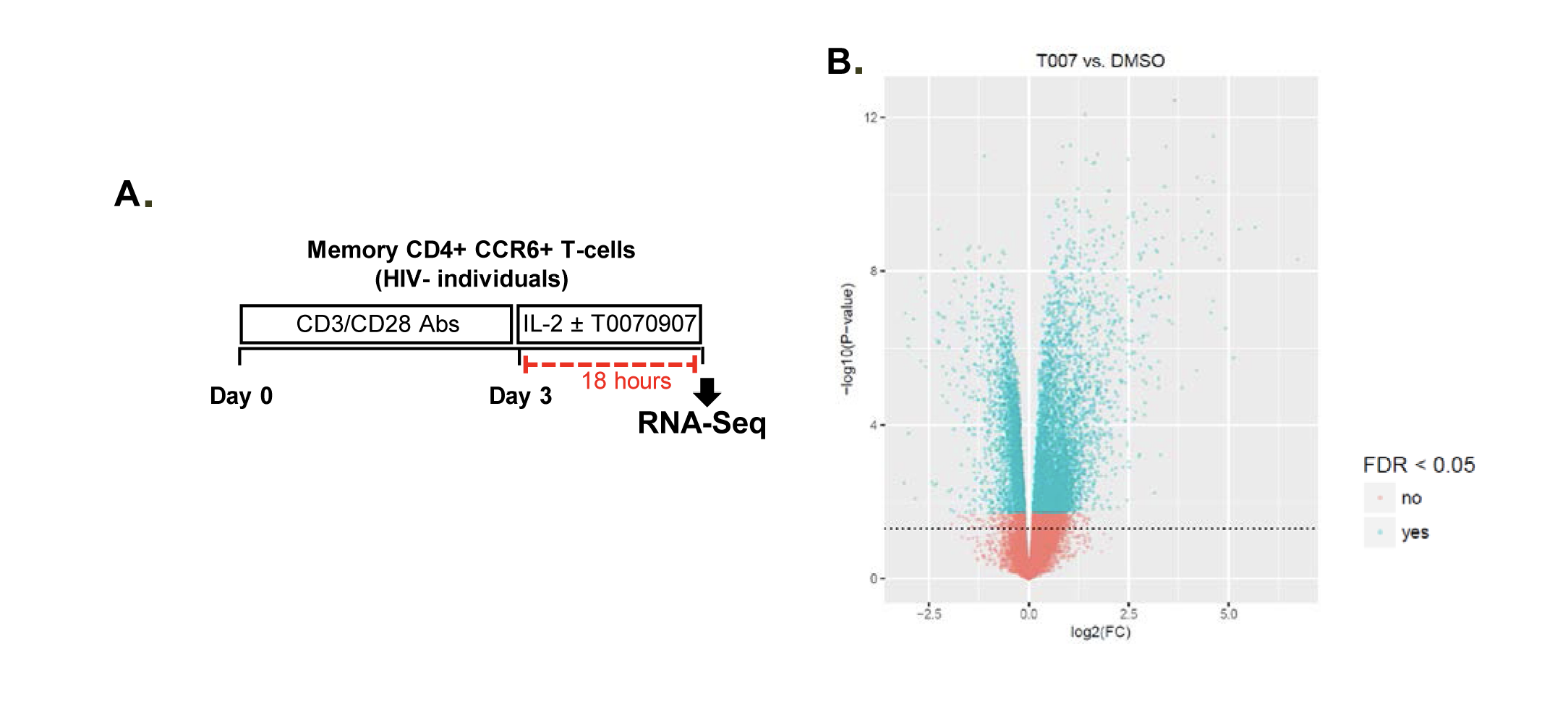
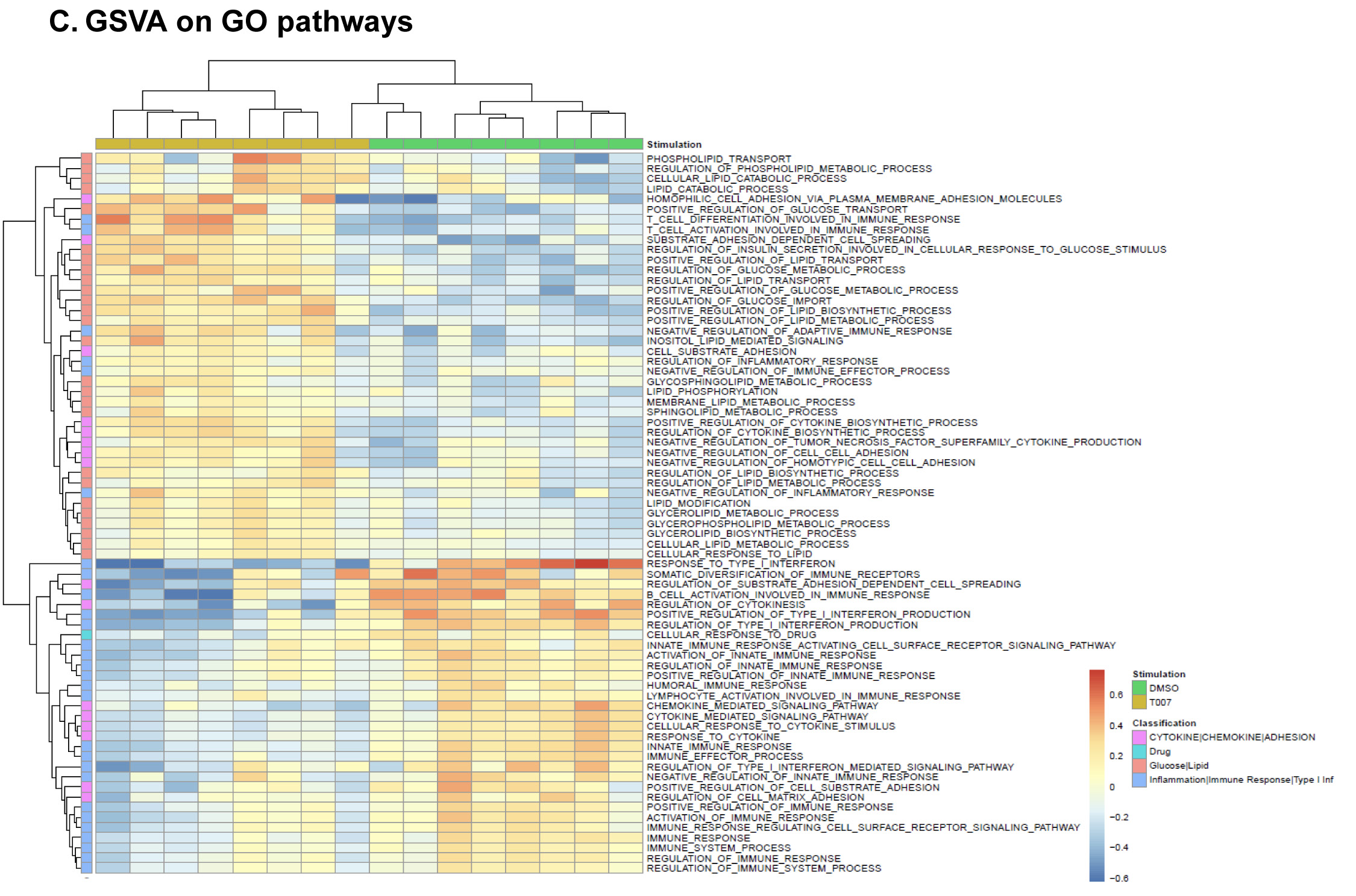
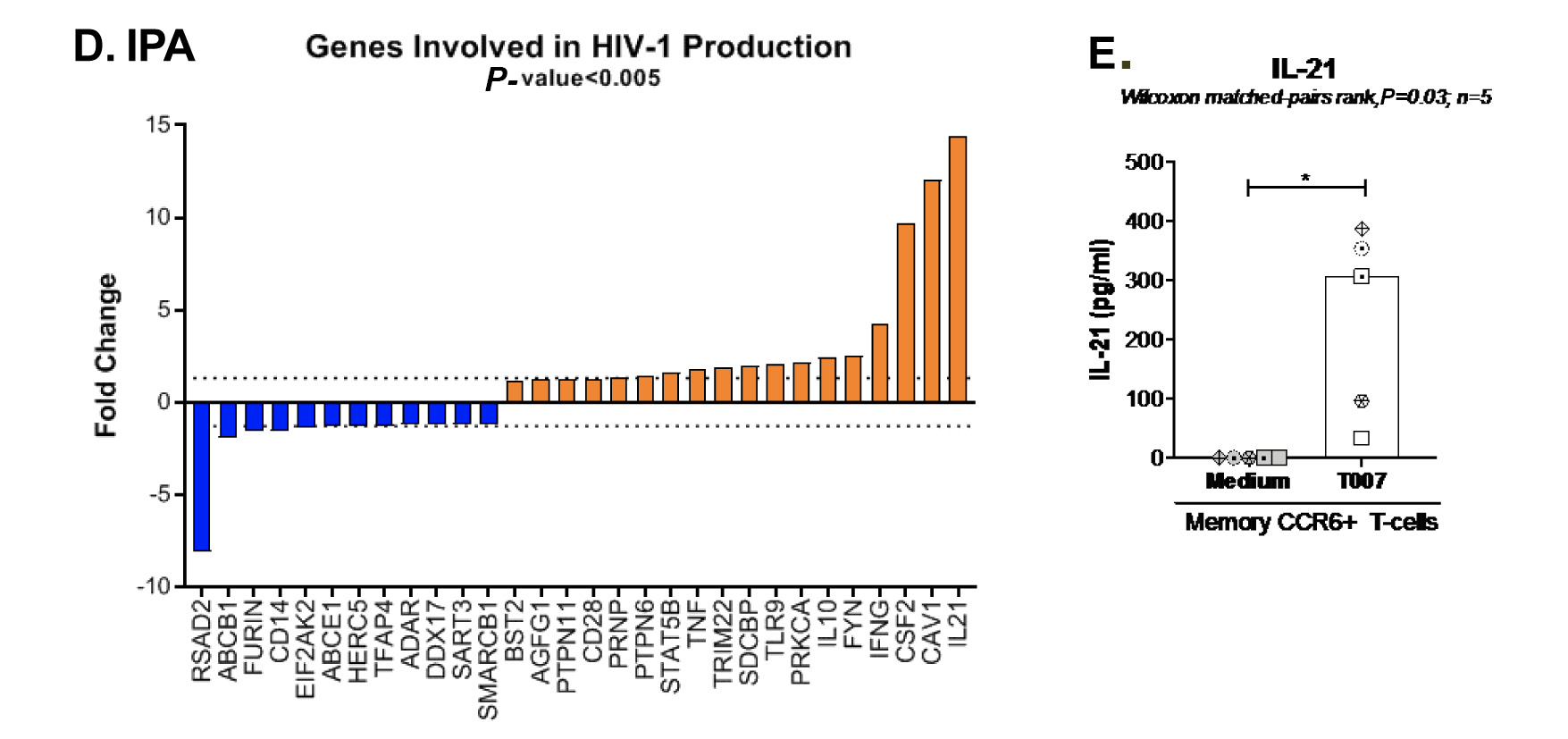
Figure 5. T0070907 imprints CCR6+CD4+ T cells with an anti-viral transcriptional program. (A) Shown is the experimental flow chart for genome-wide transcriptional analysis. Briefly, memory CCR6+ T cells of HIV-uninfected individuals (n=8) were stimulated by CD3/CD28 for 3 days and cultured with IL-2 in the presence/absence of T0070907 (10 mM) for additional 18 hours. Total RNA was extracted for RNA sequencing. (B) Volcano plots for all probes in each linear model with the log2 FC on the x-axis and the negative logarithm of the adjusted P-values for false discovery rate (FDR) on the y-axis. The red/green color code is based on the 5% FDR threshold. (C) Heatmap represents 71 pathways included in the gene ontology (GO) classification: cytokines/chemokines (pink), drug transporters (blue), glucose/lipid metabolism (orange), and inflammation/immune response to type I interferon (violet) based on the 5% FDR threshold. Heatmap cells are scaled by the expression level z-scores for each probe individually. (D) Ingenuity pathway analysis (IPA) identified genes involved in HIV-1 production and differentially modulated by T0070907 (P<0.05). The y-axis represents the FC, with the 1.3 FC cut-off indicated by the dotted line. (E) IL-21 levels in cell culture supernatants were quantified by ELISA (n=5). Each symbol represents 1 different donor; bars represent median values. Wilcoxon matched-pairs signed rank test P-value is indicated on the graph.
Gene Set Variation Analysis (GSVA) allowed the identification of Gene Ontology (GO) biological processes (false discovery rate (FDR) <0.05) using the Broad Institute data base (MSigDB C2, V6.2). Among 71 modulated pathways (Figure 5C), top pathways were linked to the GO terms: i) lipid/phospholipid and glucose metabolism (Supplementary Figure 5A-C), ii) inflammation/immune response to type I interferon (Supplementary Figure 5D), and iii) cytokines, chemokines and adhesion molecules (Supplementary Figure 5E-H). Differentially expressed genes linked to the GO term lipid/phospholipid metabolism include the upregulation of the transcription factors PPARy, PPARα, KLF4, and NR4A3; the pattern recognition receptor NOD2; the tetraspanin CD81; the signaling molecules PTK2, PLA2G6, FGF2, and FLT1; the guanine nucleotide exchange factor VAV3; the hormone ADIPOQ/adiponectin; the cytokines TNF and IFNG; the downregulation of the ATP transporter ABCG1; the G protein RAC1; and the cell cycle regulator CDC42 (Supplementary Figure 5A-B). Differentially expressed genes linked to the GO term glucose metabolism include the upregulation of the glycosylphosphatidylinositol (GPI) degrading enzyme GPLD1, the insulin-like growth factors IGF1 and IGF2, and the phorbol-12-myristate-13-acetate-induced protein 1 (PMAIP1); and the downregulation of the enzymes tyrosine-protein phosphatase non-receptor type 2 (PTPN2) and diglyceride acyltransferase (DGAT2) (Supplementary Figure 5C). Differentially expressed genes linked to the GO term inflammation/immune response to type I interferon were mainly downregulated by T0070907 and included genes documented to play a positive/negative regulatory role in HIV replication such as ADAR, MX2, MX1, OAS1, RNASEL, SAMHD1, ISG15, ISG20, IFITM2, IFITM3, and TRIM56; of note, transcripts coding for the restriction factor BST2 were upregulated (Supplementary Figure 5D). Finally, differentially expressed genes related to the GO terms cytokines, chemokines, and adhesion molecules included upregulated transcripts for chemokine receptors (CXCR5, CXCR4, CX3CR1, CCR8), chemokines (CCL20, CCL1, XCL1, XCL2), cell-to-cell adhesion molecules/immune checkpoints (CD276/B7-H3, LAG3, CTLA4, TIGIT), and cytokines/cytokine regulators (IL-4, IL-10, CD28, BCL10, STAT5B, CD3E, CD80, IL-21, KLF4, IFNG, TLR9, TNF, TNFAIP3, IRAK3, AXL, PTPN22); as well as downregulated transcripts for chemokine receptors (CCR1-3, CCR5, CCR7, CCR9, CCR10, CXCR3, CXCR6), chemokines (CCRL2), cell-to-cell adhesion molecules (CD274/PD-L1, LGALS9, CD300A, CD74, CEACAM1, TNFSF14, LGALS3, TNFSF4), and cytokine biosynthesis (TLR1, NFKB1, LTB, TLR6, NLRC3, RARA) (Supplementary Figure 5E-H).
These results reveal a previously unrecognized complex network of cellular processes that are positively or negatively controlled by PPARy in Th17-polarized CCR6+ T cells, with relevance for understanding the dichotomous effects of T0070907 on the various steps or HIV replication.
Ingenuity Pathway Analysis revealed the upregulation and downregulation of transcripts previously linked to the negative (eg, IL-21, CAV1, BST2) and positive (eg, furin) regulation of HIV replication, respectively (Figure 5D). Considering the well-documented role of IL-21 in modulating Th17/Tfh survival [14, 15], as well as its antiviral properties [44, 45, 51], we pursued the validation of IL-21 at the protein level. Results generated with memory CCR6+ T cells from 5 individuals confirmed the significant upregulation of IL-21 protein production by T0070907 (Figure 5E). IL-21 exerts its antiviral functions by the induction of miR-29 [44], a non-coding RNA that reduces HIV replication by interfering with Nef [52]. Consistently, Ingenuity Pathway Analysis (Supplementary Figure 6) revealed the interactome linked to IL-21 and the connection with miR-29 by the up-regulation of STAT3 [44]. These results point to IL-21 upregulation as one mechanism underlining the virological features of PPARy inhibition.
In addition to IL-21, T0070907 upregulated a set of Tfh-specific transcripts [53], including transcription factors (Bcl6, MAF, STAT3), chemokine receptors (CXCR4, CXCR5), surface markers (CD4, ICOS), and cytokines (IL-4, IL-10, IL-17A/F) (Supplementary Figure 7).
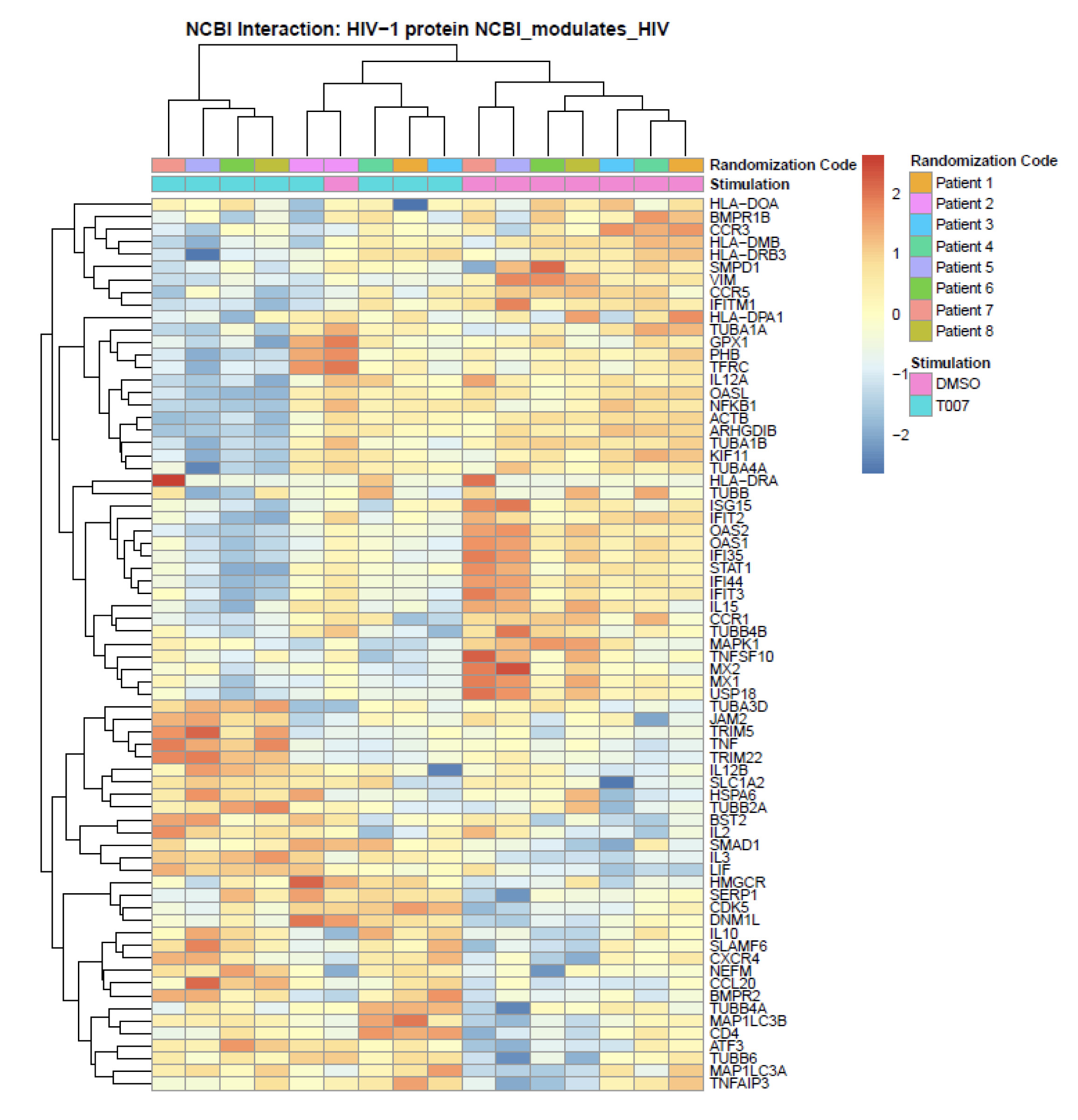
Figure 6. Meta-analysis using the NCBI HIV interaction database. Genome-wide transcriptional profiles were generated as in Figure 5. Transcripts modulated by T0070907 in CCR6+ T cells (P <0.05, FC cut-off 1.3) were matched to the lists of human genes included on the NCBI HIV interaction database. Heatmap cells are scaled by the expression level z-scores for each probe individually. Results from each donor are indicated with a different color code (n=8).
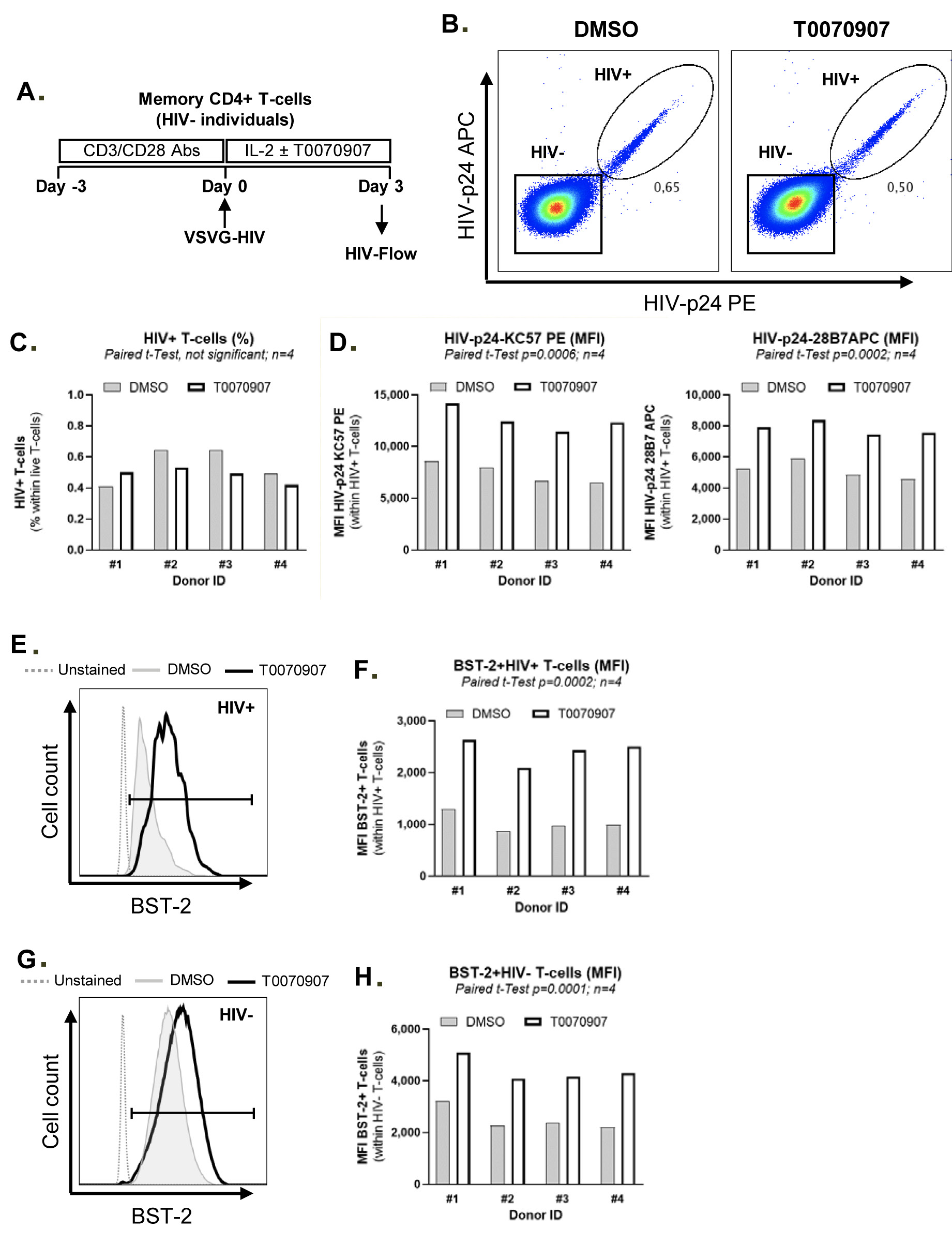
Figure 7. T0070907 prevents BST-2 downregulation on HIV-infected T cells. (A) Shown is the experimental flow chart. Briefly, memory CD4+ T cells isolated from HIV-uninfected individuals were stimulated with anti-CD3/CD28 antibodies for 3 days, exposed to single round VSV-G/HIV for 3 hours. Then, cells were cultured in the presence of IL-2 (5 ng/ml) and in the presence or the absence (DMSO) of T0070907 (10 μM) for 3 additional days. HIV-Flow using 2 distinct HIV-p24 antibody clones coupled with different fluorochromes (28B7 APC and KC57 PE), together with surface staining with BST-2 and CD4 antibodies, were performed and analyzed by flow cytometry. Shown is the co-expression of HIV-p24 PE and APC antibodies allowing the identification of productively infected cells (HIV+) in 1 representative donor (B) and the statistical analysis of the % of HIV+ cells (C) and the MFI of HIV-p24 PE and APC expression on exposure to DMSO or T0070907 in 4 different donors (D). Shown are histograms from 1 representative donor for BST-2 and CD4 expression (E and G), as well as the statistical analyses of BST-2 and CD4 expression (MFI) on HIV+ cells in 4 different donors (F and H). Paired t-test values are indicated on the graphs.
A meta-analysis using the NCBI HIV-1 interactions database allowed the identification of human genes previously involved in HIV-1 infection that are modulated by T0070907 in CCR6+ T cells. Specifically, TRIM5, TNF, TRIM22, BST2, IL-2, IL-3, LIF, IL-10, CXCR4, SERP1, and CD4 were upregulated; while VIM, CCR5, IFITM1, OASL, NFKB1, ISG15, IFIT2, OAS2, OAS1, compared to uninfected bystander ones FIT35, STAT1, IL15, MX2, MX1, and USP18 were downregulated (Figure 6). Among transcripts regulating HIV transcription, T0070907 upregulated the expression of the nuclear receptor co-activators (NCOA)1-3, the nuclear factor of activated T cells cytoplasmic 1 (NFATC1), the HIV-1 Tat Interactive Protein 2 (HTATIP2), CD3E, CD3D, IKBKB, and CDK9, and it downregulated the expression of MAPK1, NOX1, and the DNA-directed RNA polymerases POLR2C, POLR2H, POLR2D, POLR2E, POLR2F, and POLR2L (Supplementary Figure 8).
Together, these RNA-Seq results reveal that T0070907-mediated transcriptional reprogramming is associated with the negative regulation of multiple steps of the viral replication cycle such as CCR5-mediated entry, the uncoating (eg, TRIM5), reverse transcription (eg, SAMHD1), Nef-mediated functions (eg, IL-21, miR29), viral particle production (eg, TRIM22), release (eg, BST2), Env processing (eg, furin), while facilitating HIV transcription (eg, NCOA1-3, HTATIP2, CDK9), and Th17-specific effector functions (eg, RORyt, STAT3, IL-17A, IL-21).
Tetherin/BST2 represents a key HIV restriction factor downregulated by the HIV accessory protein Vpu to allow release of progeny virions from productively infected T cells [54-56]. Our RNA-Seq revealed the upregulation of BST-2 RNA in memory CCR6+ T cells on exposure to T0070907 (FC: 1.2, P=0.001; adj. P=0.007) (Figure 6, Supplementary Figures 5D and 9). The interrogation of the ENCODE dataset generated by TF ChIP-Seq analysis on HepG2 cells (https://www.encodeproject.org/experiments/ENCSR130VQL/) allowed us to identify 2,118 T0070907-modulated transcripts that are putative direct PPARy targets in T cells, with BST-2 encoding for PPREs in its promoter (data not shown). Thus, we hypothesized that increased BST-2 expression contributes to limiting release of progeny virions from infected cells, as demonstrated in Figure 1F. To test this hypothesis, we performed single-round HIV infection using a VSV-G/HIV construct (which enters cells by endocytosis independently of CD4 and co-receptors [57]), cultured cells in the presence or absence of T0070907, and analyzed by FACS the expression of BST-2 protein on the surface of HIV-infected cells identified using HIV-Flow (Figure 7A), as previously reported [58]. As expected, in the absence of T0070907, BST-2 expression was downregulated on HIV-infected T cells compared to uninfected bystander ones (Supplementary Figure 9A and C). Exposure to T0070907 led to a significant increase in the intracellular HIV-p24 expression (MFI of HIV-p24 PE and HIV-p24 APC antibody expression) (Figure 7B-D), as well as an increased BST-2 surface expression (MFI) on HIV-infected T cells (Figure 7E-F, Supplementary Figure 9" by "9A and C"). A T0070907-mediated increase in BST-2 expression was also observed on the surface of bystander HIV-uninfected T cells (Figure 7G-H, Supplementary Figure 9). These results indicate that PPARy inhibition allows an efficient HIV translation into proteins (ie, HIV-p24) and suggest that BST-2 upregulation by T0070907 contributes to limiting the release of progeny virions from productively infected T cells.
In this study, we reveal the unique features combined by the PPARy antagonist T0070907 including the positive regulation of HIV transcription/translation and Th17/Tfh-specific effector functions in memory CD4+ T cells of ART-treated PLWH, together with its capacity to reduce de novo virion production and/or spread from HIV reservoir cells. By using a genome-wide transcriptional profiling in Th17-polarized CCR6+CD4+ T cells, we revealed a complex transcriptional reprogramming underlying the observed immunological/virological features of T0070907, with antiviral mechanisms located at multiple steps of the HIV replication cycle downstream translation, including the BST-2-mediated restriction of HIV release (Figure 8).
In addition to the knowledge that PPARy acts as a repressor of HIV [31] RORgt transcription [32, 34], we demonstrate that the pharmacological inhibition of PPARy and using the antagonist T0070907 [47] boosted HIV transcription and RORgt-mediated transcription of Th17-specific genes. Conversely, we observed an unexpected blockage in the viral production and release of spread in culture as observed during viral outgrowth ex vivo and HIV infection in vitro. It is of particular importance to mention that T0070907 acted preferentially on CCR6+ Th17-polarized T cells, a subset known to be enriched in HIV reservoirs in ART-treated PLWH [16, 59], to increase IL-17A production and to reduce CCR5 expression and viral replication in vitro. Similar to T0070907, the literature documents that PKC-q activators such as prostratin, a non-tumor-promoting phorbol ester, also act as an LRA while blocking de novo HIV production to mediate the elimination of HIV reservoirs by a shock and kill strategy [60-63]. Whether the effects of prostratin and its derivatives [64] also involve PPARy-modulated processes remains to be determined. However, one major difference is that PKC-q activators downregulate CD4, while T0070907 does not.
The PPARy/RXR heterodimer is known to target genes involved in lipid metabolism such as cholesterol and fatty acids that influence multiple aspects of the antiviral immunity [26, 28, 65]. Among oxysterols presenting of the antiviral properties, 25HC, metabolized from cholesterol by the enzyme CH25H, blocks the replication of HIV by acting on the viral entry but not on the transcription [66], with the effects on the post-transcriptional steps of the replication cycle remaining unexplored. In addition, 25HC has been identified as a natural ligand for RORyt [67, 68]. The fact that PPARy deficiency was linked to CH25H overexpression [69] prompted our initial hypothesis that T0070907 blocks HIV outgrowth ex vivo and infection in vitro and boosts Th17 effector functions via CH25H/25HC-dependent mechanisms. In confirmation of this, T0070907 upregulates the expression of CH25H mRNA in TCR-activated CCR6- non-Th17 cells (data not shown), further explaining their relative resistance to HIV infection [14, 15]. However, CH25H mRNA was undetectable in CCR6+ Th17 cells (data not shown), indicating that T0070907 exerts its antiviral effects in Th17 cells via CH25H/25HC-independent mechanisms.
To investigate mechanisms by which T0070907 disconnects HIV transcription from downstream viral replication steps, we performed a genome-wide transcriptional profiling using the RNA-Seq Illumina technology. GSVA identified activation of pathways linked to lipid/phospholipid and glucose metabolism. Metabolic reprogramming during TCR triggering trains T cells to integrate immunological and metabolic information required for the subsequent acquisition of specific effector functions [70]. Glucose metabolism has been identified to play a central role in HIV replication, with the glucose transporter GLUT1 being a marker for HIV permissive T cells [71]. Metabolism disruption is associated with HIV disease progression, with higher glucose uptake being observed in CD4+ T cells of PLWH compared to non-infected individuals [72]. Recent studies linked the susceptibility to HIV infection to the metabolic status of specific CD4+ T cell subsets [73]. Changes in the CD4+ T cell metabolic program are controlled by the mTORC1/PPARy axis [70, 74]. In line with this, T0070907 upregulated genes associated with PI3K/Akt signaling, a pathway known to promote mTOR activation [15]. Indeed, several groups including ours, identified mTOR as a positive regulator of HIV replication [20], acting at the level of viral entry [75] and transcription [76, 77]. Indeed, in preliminary studies, we demonstrated that TCR triggering in the presence of T0070907 leads to increased mTOR phosphorylation. Therefore, the activation of the PI3K/Akt pathway in the presence of T0070907 might be in part responsible of the increase in HIV transcription, likely via mTOR-dependent mechanisms.
GSVA identified pathways modulated by T0070907 in CCR6+ T cells revealed that PPARy antagonism produces profound transcriptional modifications linked to the metabolism of cellular membrane components, including glycosaminoglycan, glycosphingolipid, and sphingolipid. These components of the cellular membrane play a key role in the membrane organization and membrane raft formations [29]. Membrane receptors such as the HIV co-receptors CCR5/CXCR4 are recruited to the membrane rafts, and the clustering of these receptors promotes HIV entry into target cells [78]. In addition, membrane rafts play a crucial role in HIV-1 assembly and release [79, 80]. Therefore, modification of the cellular composition and membrane raft formation by T0070907 may contribute to the decreased HIV entry/release; additional investigations are needed to clarify this. The formation of biofilms rich in collagen and cell-host molecules such as tetherin/BST2 has been reported for human T cell leukemia virus type 1 (HTLV-1) [81]. The possibility that other viruses such as HIV form biofilms remains to be determined [82]. Of note, the main upregulated gene by T0070907 is fibromodulin (FMOD), a component of the extracellular matrix which participates in the assembly of collagen fibers. In line with this, the collagen triple helix repeat containing 1 (CTHRC1) and the tetherin/BST2 transcripts were upregulated by T0070907. These findings suggest that T0070907 likely facilitates the establishment of biofilms able to trap newly produced virions thus preventing their spreading.
The GSVA of GO pathways also revealed the downregulation of pathways/transcripts linked to interferon responses. Multiple interferon-stimulated genes (ISG), documented to restrict HIV replication, were downregulated by T0070907 in CCR6+ T cells. Among these transcripts, we noted a decreased expression of SAMHD1, which limits HIV reverse transcription and promotes HIV-RNA degradation [83]; MX2, which limits viral decapsidation, pre-integration complex formation and nuclear import [84, 85]; IFITM2 and IFITM3, known to interact with HIV-1 Env in infected cells and to impair Env processing and incorporation into virions [86]; and ISG15, known to induce ISGylation of viral Gag proteins and to impeded HIV release [87]. These results point to a previously unrecognized implication of PPARy in the positive transcriptional regulation of specific HIV-restriction factors, including SAMHD1, MX2, IFITM2, IFITM3, BST2, and ISG15, in line with the antiviral program promoted by PPARy activation [17].
Our RNA-Seq results also revealed a T0070907-mediated increase in the expression of the classical Tfh markers CXCR5, ICOS, BCL6, PD-1, CD40L, IL-10, and IL-21. In line with this, previous investigations demonstrated that PPARy activation prevents Tfh differentiation [33]. Of note, by boosting IL-21 production, T0070907 may improve Th17/Tfh survival and their effector functions. Indeed, in a model of SIV infection, the IL-21 supplementation of ART reduced inflammation, restored mucosal Th17 frequency, decreased the size of viral reservoir [45, 46], and also delayed viral rebound on ART interruption [45]. In addition, IL-21 exhibited antiviral functions by the induction of miR-29 [44] that targeted HIV-Nef for degradation [52]. The IL-21/miR-29 axis was also linked to a slow-down of HIV disease progression [51]. Therefore, the IL-21/miR-29 axis is highly likely to contribute to the antiviral effects of PPARy antagonism.
The meta-analysis performed using the NCBI HIV-1 interaction database pointed to additional T0070907-mediated antiviral mechanisms. Specifically, T0070907 upregulated expression of CAV1, reported to inhibit HIV particle production in macrophages [88]; SERINC5, which is incorporated into virions and prevents the fusion of the virion with the cellular membrane of a new target cell [89]; TRIM22, which blocks Gag migration to the plasma membrane and inhibits HIV particle production [90]; and BST2, which limits viral particle release [83]. A T0070907-mediated upregulation of the HIV restriction factor TRIM5α, which interacts with the HIV capsid and induces its proteasomal degradation leading to premature decapsidation [91], was also observed. Finally, T0070907 downregulated furin, a protease preferentially expressed in Th17 cells [17, 18] and involved in HIV protein Env maturation and virion infectivity [92]. Thus, the antiviral features of T0070907 involve mechanisms dependent on CAV1, SERINC5, TRIM22, and BST2 over-expression, as well as furin downregulation, thus explaining a post-transcriptional blockage in HIV virion production and/or release.
Finally, the counterintuitive capacity of PPARy antagonism to decrease viral release/outgrowth while increasing viral transcription prompted us to focus on Tetherin/BST-2, an HIV restriction factor counteracted by Vpu and documented to mediate HIV tethering on the surface of infected cells [54-56]. Of note, T0070907 increased BST-2 mRNA expression in uninfected CCR6+CD4+ T cells. In a model of single round VSV-G/HIV infection in vitro, as expected, BST-2 protein expression was downregulated on infected T cells in the absence of T0070907. In contrast, the BST-2 expression was significantly higher on the surface of infected cells exposed to T0070907. An in silico search using the ENCODE database revealed that BST-2 encodes PPREs in its promoter and represents a putative direct PPARy target in CD4+ T cells. Thus, PPARy inhibition boosts HIV reactivation, while preventing progeny virion release from infected cells via BST-2-dependent mechanisms. The recognition of such reactivated viral reservoirs by antibodies and immune cells for subsequent clearance will be key for HIV cure. Future studies in vitro and in preclinical models are needed to determine whether PPARy antagonism promotes HIV reservoir purging in shock and kill strategies.
In conclusion, our results reveal complex previously unrecognized PPARy-dependent host-cell molecular circuits involved in the positive, as well as the negative regulation of various steps of the HIV replication cycle and demonstrate the possibility of disconnecting HIV transcription and translation from viral particle production/release (Figure 8). The efficacy of the PPARy antagonism in boosting IL-21 production is of major importance, considering IL-21 paucity during HIV infection [14, 15] and its documented antiviral/immune-regulatory features [44-46, 51]. Therefore, the pharmacological inhibition of PPARy may represent a new promising therapeutic strategy to boost Th17-effector functions that are key for mucosal immunity restoration and to promote HIV-reservoir purging in ART-treated PLWH.
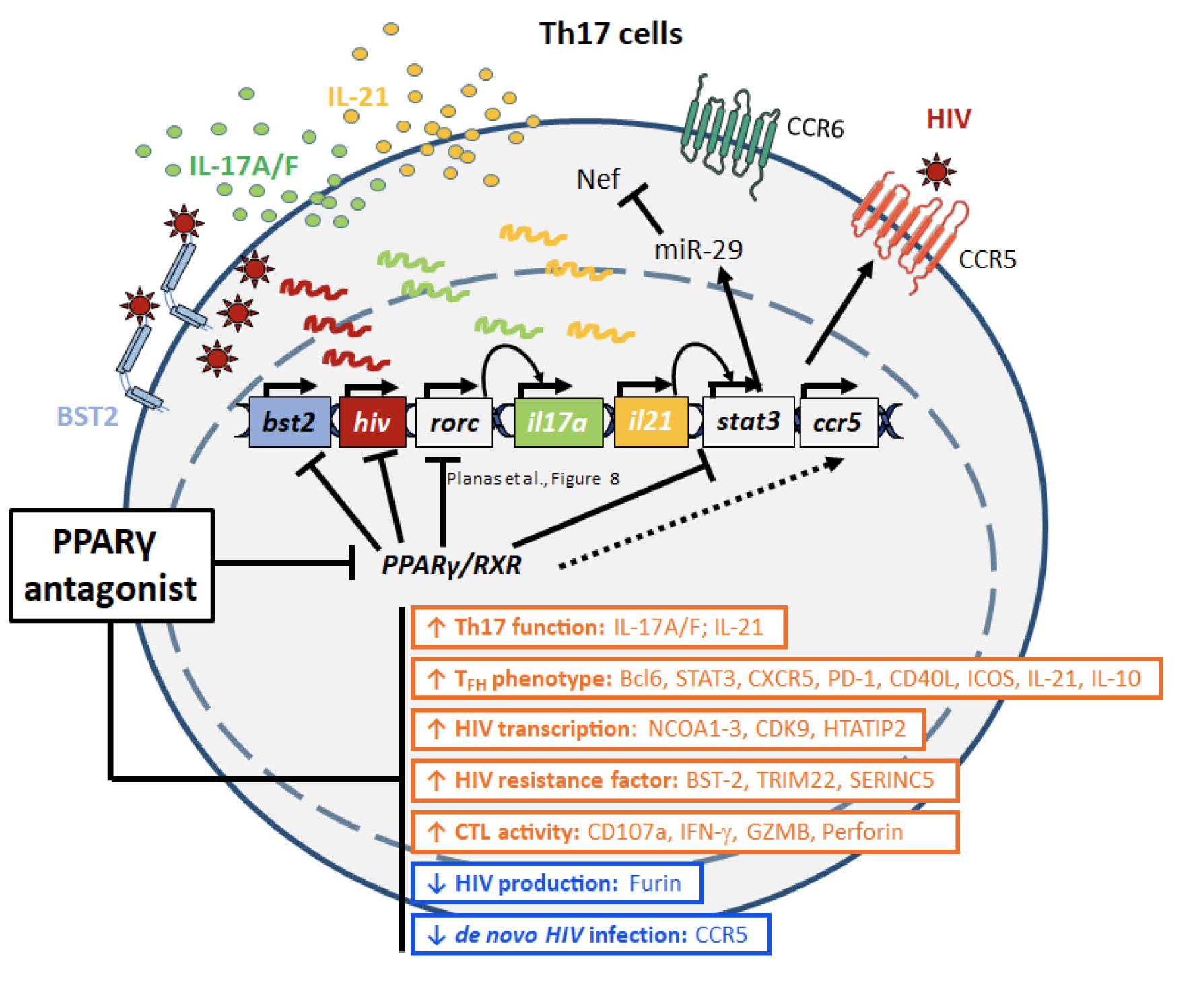
Figure 8. Summary of PPARy antagonism-mediated virological/immunological reprogramming of CCR6+ T cells. In line with the documented capacity of PPARy to repress HIV and RORgt transcription, the PPARy antagonist T0070907 acted on CCR6+ Th17 cells to boost both HIV transcription (NCOA1-3, HTATIP2, CDK9) and the expression of specific Th17/Tfh transcripts (eg, IL-17A, IL-21). Unexpectedly, the PPARy antagonism prevented de novo production/release of virions from reservoir cells by negatively interfering with multiple steps of the HIV replication cycle, from virion maturation (eg, furin) and viral particle release (eg, BST2), to viral entry into new target cells (eg, CCR5), as well as the IL-21/miR-29 antiviral axis. Thus, the PPARy antagonism may represent a new strategy to eradicate HIV reservoirs in Th17 cells.
DP, AF, and YZ designed and performed research, analyzed data, and wrote the manuscript. JPG performed RNA-Seq analysis and prepared figures. JR and AF designed and performed research, analyzed data and prepared figures. MJR, LRM, DC, HC, TRWS, and AG performed research and contributed to biological sample collection/preparation. EAC provided expertise with virological assays and contributed to manuscript writing. JPR allowed access to biological samples and study participant clinical information. NC provided expertise, protocols, and reagents, contributed to research design and manuscript writing. PA conceived the research study hypothesis, designed research, analyzed data, and wrote the manuscript.
DP, AF, YZ, JPG, JR, AF, MJR, LRM, DC, HC, TRWS, and AG declare no financial or non-financial competing interests to disclose.
EAC is a member of the Scientific Advisory Board of Theratechnologies.
JPR performed contract research and/or served on Advisory Boards for Gilead Sciences Canada Inc., Merck Canada Inc., Abbvie Corp., ViiV Healthcare, Bristol Myers Squibb, Janssen Inc., Argos Pharmaceuticals from InnaVirVax, and Theravectys.
NC received research funding from EMD Serono and served on Advisory Boards for Gilead Sciences.
PA’s laboratory receives research funding from Glaxo Smith Klein/NeoMed for projects different from the present study. PA served as a Consultant at Merck Canada Inc.
The authors thank Dr. Dominique Gauchat and Philippe St Onge (Flow Cytometry Core Facility, CHUM-Research Center, Montréal, QC, Canada) for expert technical support with polychromatic flow cytometry sorting. The authors thank Olfa Debbeche (NLC3 Core Facility CHUM-Research Center, Montréal, QC, Canada); Mario Legault for helping with ethical approvals and informed consents, and Josée Girouard, Angie Massicotte, and Cynthia Dion, for their key contribution to study participant recruitment and for access to blood samples and clinical information from HIV-infected and uninfected donors. The authors thank Dr. Robert Lodge for providing protocols and valuable experimental input. The authors thank Wilfried-Wenceslas Bazie and Dr. Caroline Gilbert (Centre de recherche du CHUL, Québec, QC, Canada) for their valuable experimental input. The authors address a special thanks to all PLWH study participants for their key contribution to this work.
1. Barre-Sinoussi F, Ross AL, Delfraissy JF. Past, present and future: 30 years of HIV research. Nature reviews Microbiology. 2013;11(12):877-83. Epub 2013/10/29. doi: 10.1038/nrmicro3132. PubMed PMID: 24162027.
2. Martin AR, Siliciano RF. Progress Toward HIV Eradication: Case Reports, Current Efforts, and the Challenges Associated with Cure. Annual review of medicine. 2016;67:215-28. Epub 2015/11/04. doi: 10.1146/annurev-med-011514-023043. PubMed PMID: 26526767.
3. Deeks SG, Lewin SR, Ross AL, Ananworanich J, Benkirane M, Cannon P, Chomont N, Douek D, Lifson JD, Lo YR, Kuritzkes D, Margolis D, Mellors J, Persaud D, Tucker JD, Barre-Sinoussi F, International ASTaCWG, Alter G, Auerbach J, Autran B, Barouch DH, Behrens G, Cavazzana M, Chen Z, Cohen EA, Corbelli GM, Eholie S, Eyal N, Fidler S, Garcia L, Grossman C, Henderson G, Henrich TJ, Jefferys R, Kiem HP, McCune J, Moodley K, Newman PA, Nijhuis M, Nsubuga MS, Ott M, Palmer S, Richman D, Saez-Cirion A, Sharp M, Siliciano J, Silvestri G, Singh J, Spire B, Taylor J, Tolstrup M, Valente S, van Lunzen J, Walensky R, Wilson I, Zack J. International AIDS Society global scientific strategy: towards an HIV cure 2016. Nature medicine. 2016;22(8):839-50. Epub 2016/07/12. doi: 10.1038/nm.4108. PubMed PMID: 27400264; PMCID: PMC5322797.
4. Chomont N, DaFonseca S, Vandergeeten C, Ancuta P, Sekaly RP. Maintenance of CD4+ T-cell memory and HIV persistence: keeping memory, keeping HIV. Curr Opin HIV AIDS. 2011;6(1):30-6. Epub 2011/01/19. doi: 10.1097/COH.0b013e3283413775. PubMed PMID: 21242891.
5. Sengupta S, Siliciano RF. Targeting the Latent Reservoir for HIV-1. Immunity. 2018;48(5):872-95. Epub 2018/05/17. doi: 10.1016/j.immuni.2018.04.030. PubMed PMID: 29768175; PMCID: PMC6196732.
6. Colby DJ, Trautmann L, Pinyakorn S, Leyre L, Pagliuzza A, Kroon E, Rolland M, Takata H, Buranapraditkun S, Intasan J, Chomchey N, Muir R, Haddad EK, Tovanabutra S, Ubolyam S, Bolton DL, Fullmer BA, Gorelick RJ, Fox L, Crowell TA, Trichavaroj R, O’Connell R, Chomont N, Kim JH, Michael NL, Robb ML, Phanuphak N, Ananworanich J, group RVs. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nature medicine. 2018;24(7):923-6. Epub 2018/06/13. doi: 0.1038/s41591-018-0026-61. PubMed PMID: 29892063; PMCID: PMC6092240.
7. El-Far M, Tremblay CL. Gut microbial diversity in HIV infection post combined antiretroviral therapy: a key target for prevention of cardiovascular disease. Curr Opin HIV AIDS. 2018;13(1):38-44. Epub 2017/10/19. doi: 10.1097/COH.0000000000000426. PubMed PMID: 29045253; PMCID: PMC5718258.
8. Paul R. Neurocognitive Phenotyping of HIV in the Era of Antiretroviral Therapy. Curr HIV/AIDS Rep. 2019;16(3):230-5. Epub 2019/06/07. doi: 10.1007/s11904-019-00426-9. PubMed PMID: 31168712; PMCID: PMC6571160.
9. Davenport MP, Khoury DS, Cromer D, Lewin SR, Kelleher AD, Kent SJ. Functional cure of HIV: the scale of the challenge. Nat Rev Immunol. 2019;19(1):45-54. Epub 2018/11/10. doi: 10.1038/s41577-018-0085-4. PubMed PMID: 30410126.
10. Bruner KM, Hosmane NN, Siliciano RF. Towards an HIV-1 cure: measuring the latent reservoir. Trends in microbiology. 2015;23(4):192-203. Epub 2015/03/10. doi: 10.1016/j.tim.2015.01.013. PubMed PMID: 25747663; PMCID: PMC4386620.
11. Kuo HH, Lichterfeld M. Recent progress in understanding HIV reservoirs. Curr Opin HIV AIDS. 2018;13(2):137-42. Epub 2017/12/13. doi: 10.1097/COH.0000000000000441. PubMed PMID: 29232209; PMCID: PMC5806203.
12. Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy JP, Haddad EK, Sekaly RP. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nature medicine. 2009;15(8):893-900. Epub 2009/06/23. doi: 10.1038/nm.1972. PubMed PMID: 19543283; PMCID: PMC2859814.
13. Clayton KL, Garcia JV, Clements JE, Walker BD. HIV Infection of Macrophages: Implications for Pathogenesis and Cure. Pathog Immun. 2017;2(2):179-92. Epub 2017/07/29. doi: 10.20411/pai.v2i2.204. PubMed PMID: 28752134; PMCID: PMC5526341.
14. Wacleche VS, Landay A, Routy JP, Ancuta P. The Th17 Lineage: From Barrier Surfaces Homeostasis to Autoimmunity, Cancer, and HIV-1 Pathogenesis. Viruses. 2017;9(10). Epub 2017/10/20. doi: 10.3390/v9100303. PubMed PMID: 29048384; PMCID: PMC5691654.
15. Planas D, Routy JP, Ancuta P. New Th17-specific therapeutic strategies for HIV remission. Curr Opin HIV AIDS. 2019;14(2):85-92. Epub 2018/12/14. doi: 10.1097/COH.0000000000000522. PubMed PMID: 30543544.
16. Anderson JL, Khoury G, Fromentin R, Solomon A, Chomont N, Sinclair E, Milush JM, Hartogensis W, Bacchetti P, Roche M, Tumpach C, Gartner M, Pitman MC, Epling CL, Hoh R, Hecht FM, Somsouk M, Cameron PU, Deeks SG, Lewin SR. Human Immunodeficiency Virus (HIV)-Infected CCR6+ Rectal CD4+ T Cells and HIV Persistence On Antiretroviral Therapy. J Infect Dis. 2020;221(5):744-55. Epub 2019/12/05. doi: 10.1093/infdis/jiz509. PubMed PMID: 31796951; PMCID: PMC7026892.
17. Bernier A, Cleret-Buhot A, Zhang Y, Goulet JP, Monteiro P, Gosselin A, DaFonseca S, Wacleche VS, Jenabian MA, Routy JP, Tremblay C, Ancuta P. Transcriptional profiling reveals molecular signatures associated with HIV permissiveness in Th1Th17 cells and identifies peroxisome proliferator-activated receptor gamma as an intrinsic negative regulator of viral replication. Retrovirology. 2013;10:160. Epub 2013/12/24. doi: 10.1186/1742-4690-10-160. PubMed PMID: 24359430; PMCID: PMC3898812.
18. Cleret-Buhot A, Zhang Y, Planas D, Goulet JP, Monteiro P, Gosselin A, Wacleche VS, Tremblay CL, Jenabian MA, Routy JP, El-Far M, Chomont N, Haddad EK, Sekaly RP, Ancuta P. Identification of novel HIV-1 dependency factors in primary CCR4(+)CCR6(+)Th17 cells via a genome-wide transcriptional approach. Retrovirology. 2015;12(1):102. Epub 2015/12/15. doi: 10.1186/s12977-015-0226-9. PubMed PMID: 26654242; PMCID: PMC4676116.
19. Wacleche VS, Goulet JP, Gosselin A, Monteiro P, Soudeyns H, Fromentin R, Jenabian MA, Vartanian S, Deeks SG, Chomont N, Routy JP, Ancuta P. New insights into the heterogeneity of Th17 subsets contributing to HIV-1 persistence during antiretroviral therapy. Retrovirology. 2016;13(1):59. Epub 2016/08/25. doi: 10.1186/s12977-016-0293-6. PubMed PMID: 27553844; PMCID: PMC4995622.
20. Planas D, Zhang Y, Monteiro P, Goulet JP, Gosselin A, Grandvaux N, Hope TJ, Fassati A, Routy JP, Ancuta P. HIV-1 selectively targets gut-homing CCR6+CD4+ T cells via mTOR-dependent mechanisms. JCI Insight. 2017;2(15). Epub 2017/08/05. doi: 10.1172/jci.insight.93230. PubMed PMID: 28768913; PMCID: PMC5543920.
21. Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2(10):748-59. Epub 2002/10/03. doi: 10.1038/nri912. PubMed PMID: 12360213.
22. Potula R, Ramirez SH, Knipe B, Leibhart J, Schall K, Heilman D, Morsey B, Mercer A, Papugani A, Dou H, Persidsky Y. Peroxisome proliferator-activated receptor-gamma activation suppresses HIV-1 replication in an animal model of encephalitis. AIDS. 2008;22(13):1539-49. Epub 2008/08/02. doi: 10.1097/QAD.0b013e3283081e08. PubMed PMID: 18670212; PMCID: PMC2688810.
23. Skolnik PR, Rabbi MF, Mathys JM, Greenberg AS. Stimulation of peroxisome proliferator-activated receptors alpha and gamma blocks HIV-1 replication and TNFalpha production in acutely infected primary blood cells, chronically infected U1 cells, and alveolar macrophages from HIV-infected subjects. J Acquir Immune Defic Syndr. 2002;31(1):1-10. Epub 2002/09/28. doi: 10.1097/00126334-200209010-00001. PubMed PMID: 12352144.
24. Hanley TM, Blay Puryear W, Gummuluru S, Viglianti GA. PPARgamma and LXR signaling inhibit dendritic cell-mediated HIV-1 capture and trans-infection. PLoS Pathog. 2010;6:e1000981. Epub 2010/07/10. doi: 10.1371/journal.ppat.1000981. PubMed PMID: 20617179; PMCID: PMC2895661.
25. Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature. 2008;456(7220):350-6. Epub 2008/12/02. doi: 10.1038/nature07413. PubMed PMID: 19043829; PMCID: PMC2743566.
26. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARgamma signaling and metabolism: the good, the bad and the future. Nature medicine. 2013;19(5):557-66. Epub 2013/05/09. doi: 10.1038/nm.3159. PubMed PMID: 23652116; PMCID: PMC3870016.
27. Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10(5):365-76. Epub 2010/04/24. doi: 10.1038/nri2748. PubMed PMID: 20414208.
28. Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nature reviews Cancer. 2012;12(3):181-95. Epub 2012/02/10. doi: 10.1038/nrc3214. PubMed PMID: 22318237; PMCID: PMC3322353.
29. Robinson GA, Waddington KE, Pineda-Torra I, Jury EC. Transcriptional Regulation of T-Cell Lipid Metabolism: Implications for Plasma Membrane Lipid Rafts and T-Cell Function. Front Immunol. 2017;8:1636. Epub 2017/12/12. doi: 10.3389/fimmu.2017.01636. PubMed PMID: 29225604; PMCID: PMC5705553.
30. Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, Yokote K, Nakayama T. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun. 2016;7:13683. Epub 2016/12/03. doi: 10.1038/ncomms13683. PubMed PMID: 27901044; PMCID: PMC5141517.
31. Ladias JA. Convergence of multiple nuclear receptor signaling pathways onto the long terminal repeat of human immunodeficiency virus-1. J Biol Chem. 1994;269(8):5944-51. Epub 1994/02/25. PubMed PMID: 8119938.
32. Klotz L, Knolle P. Nuclear receptors: TH17 cell control from within. FEBS Lett. 2011;585(23):3764-9. Epub 2011/07/13. doi: 10.1016/j.febslet.2011.06.027. PubMed PMID: 21745474.
33. Park HJ, Kim DH, Choi JY, Kim WJ, Kim JY, Senejani AG, Hwang SS, Kim LK, Tobiasova Z, Lee GR, Craft J, Bothwell AL, Choi JM. PPARgamma negatively regulates T cell activation to prevent follicular helper T cells and germinal center formation. PLoS One. 2014;9(6):e99127. Epub 2014/06/13. doi: 10.1371/journal.pone.0099127. PubMed PMID: 24921943; PMCID: PMC4055678.
34. Klotz L, Burgdorf S, Dani I, Saijo K, Flossdorf J, Hucke S, Alferink J, Nowak N, Beyer M, Mayer G, Langhans B, Klockgether T, Waisman A, Eberl G, Schultze J, Famulok M, Kolanus W, Glass C, Kurts C, Knolle PA. The nuclear receptor PPAR gamma selectively inhibits Th17 differentiation in a T cell-intrinsic fashion and suppresses CNS autoimmunity. J Exp Med. 2009;206(10):2079-89. Epub 2009/09/10. doi: 10.1084/jem.20082771. PubMed PMID: 19737866; PMCID: PMC2757877.
35. Hadigan C. Peroxisome proliferator-activated receptor gamma agonists and the treatment of HIV-associated lipoatrophy: unraveling the molecular mechanism of their shortcomings. J Infect Dis. 2008;198(12):1729-31. Epub 2008/10/29. doi: 10.1086/593180. PubMed PMID: 18954262.
36. Hadigan C, Mazza S, Crum D, Grinspoon S. Rosiglitazone increases small dense low-density lipoprotein concentration and decreases high-density lipoprotein particle size in HIV-infected patients. AIDS. 2007;21(18):2543-6. Epub 2007/11/21. doi: 10.1097/QAD.0b013e3282f25123. PubMed PMID: 18025896; PMCID: PMC4391618.
37. Mulligan K, Yang Y, Wininger DA, Koletar SL, Parker RA, Alston-Smith BL, Schouten JT, Fielding RA, Basar MT, Grinspoon S. Effects of metformin and rosiglitazone in HIV-infected patients with hyperinsulinemia and elevated waist/hip ratio. AIDS. 2007;21(1):47-57. Epub 2006/12/07. doi: 10.1097/QAD.0b013e328011220e. PubMed PMID: 17148967.
38. Leung V, Chiu YL, Kotler DP, Albu J, Zhu YS, Ham K, Engelson ES, Hammad H, Christos P, Donovan DS, Ginsberg HN, Glesby MJ. Effect of Recombinant Human Growth Hormone and Rosiglitazone for HIV-Associated Abdominal Fat Accumulation on Adiponectin and other Markers of Inflammation. HIV Clin Trials. 2016;17(2):55-62. Epub 2016/04/15. doi: 10.1080/15284336.2015.1126424. PubMed PMID: 27077672; PMCID: PMC4941209.
39. Tungsiripat M, El-Bejjani D, Rizk N, Dogra V, O’Riordan MA, Ross AC, Hileman C, Storer N, Harrill D, McComsey GA. Carotid intima media thickness, inflammatory markers, and endothelial activation markers in HIV Patients with lipoatrophy increased at 48 weeks regardless of use of rosiglitazone or placebo. AIDS Res Hum Retroviruses. 2011;27(3):295-302. Epub 2010/10/26. doi: 10.1089/aid.2010.0187. PubMed PMID: 20969457; PMCID: PMC3064528.
40. Prost S, Le Dantec M, Auge S, Le Grand R, Derdouch S, Auregan G, Deglon N, Relouzat F, Aubertin AM, Maillere B, Dusanter-Fourt I, Kirszenbaum M. Human and simian immunodeficiency viruses deregulate early hematopoiesis through a Nef/PPARgamma/STAT5 signaling pathway in macaques. J Clin Invest. 2008;118(5):1765-75. Epub 2008/04/24. doi: 10.1172/JCI33037. PubMed PMID: 18431514; PMCID: PMC2323187.
41. Kirchhoff F, Silvestri G. Is Nef the elusive cause of HIV-associated hematopoietic dysfunction? J Clin Invest. 2008;118(5):1622-5. Epub 2008/04/24. doi: 10.1172/JCI35487. PubMed PMID: 18431512; PMCID: PMC2323195.
42. Margolis DM, Archin NM. Proviral Latency, Persistent Human Immunodeficiency Virus Infection, and the Development of Latency Reversing Agents. J Infect Dis. 2017;215(suppl_3):S111-S8. Epub 2017/05/19. doi: 10.1093/infdis/jiw618. PubMed PMID: 28520964; PMCID: PMC5410986.
43. Kim Y, Anderson JL, Lewin SR. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe. 2018;23(1):14-26. Epub 2018/01/13. doi: 10.1016/j.chom.2017.12.004. PubMed PMID: 29324227; PMCID: PMC5990418.
44. Adoro S, Cubillos-Ruiz JR, Chen X, Deruaz M, Vrbanac VD, Song M, Park S, Murooka TT, Dudek TE, Luster AD, Tager AM, Streeck H, Bowman B, Walker BD, Kwon DS, Lazarevic V, Glimcher LH. IL-21 induces antiviral microRNA-29 in CD4 T cells to limit HIV-1 infection. Nat Commun. 2015;6:7562. Epub 2015/06/26. doi: 10.1038/ncomms8562. PubMed PMID: 26108174; PMCID: PMC4481879.
45. Micci L, Ryan ES, Fromentin R, Bosinger SE, Harper JL, He T, Paganini S, Easley KA, Chahroudi A, Benne C, Gumber S, McGary CS, Rogers KA, Deleage C, Lucero C, Byrareddy SN, Apetrei C, Estes JD, Lifson JD, Piatak M, Jr., Chomont N, Villinger F, Silvestri G, Brenchley JM, Paiardini M. Interleukin-21 combined with ART reduces inflammation and viral reservoir in SIV-infected macaques. J Clin Invest. 2015;125(12):4497-513. Epub 2015/11/10. doi: 10.1172/JCI81400. PubMed PMID: 26551680; PMCID: PMC4665780.
46. Pallikkuth S, Micci L, Ende ZS, Iriele RI, Cervasi B, Lawson B, McGary CS, Rogers KA, Else JG, Silvestri G, Easley K, Estes JD, Villinger F, Pahwa S, Paiardini M. Maintenance of intestinal Th17 cells and reduced microbial translocation in SIV-infected rhesus macaques treated with interleukin (IL)-21. PLoS Pathog. 2013;9(7):e1003471. Epub 2013/07/16. doi: 10.1371/journal.ppat.1003471. PubMed PMID: 23853592; PMCID: PMC3701718.
47. Vandergeeten C, Fromentin R, DaFonseca S, Lawani MB, Sereti I, Lederman MM, Ramgopal M, Routy JP, Sekaly RP, Chomont N. Interleukin-7 promotes HIV persistence during antiretroviral therapy. Blood. 2013;121(21):4321-9. Epub 2013/04/17. doi: 10.1182/blood-2012-11-465625. PubMed PMID: 23589672; PMCID: PMC3663425.
48. Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, Hahn BH, Kappes JC. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J Virol. 2012;86(5):2715-28. Epub 2011/12/23. doi: 10.1128/JVI.06157-11. PubMed PMID: 22190722; PMCID: PMC3302286.
49. Ancuta P, Kunstman KJ, Autissier P, Zaman T, Stone D, Wolinsky SM, Gabuzda D. CD16+ monocytes exposed to HIV promote highly efficient viral replication upon differentiation into macrophages and interaction with T cells. Virology. 2006;344(2):267-76. Epub 2005/11/25. doi: XXX10.1016/j.virol.2005.10.027. PubMed PMID: 16305804.
50. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34(5):525-7. Epub 2016/04/05. doi: 10.1038/nbt.3519. PubMed PMID: 27043002.
51. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. Epub 2015/01/22. doi: 10.1093/nar/gkv007. PubMed PMID: 25605792; PMCID: PMC4402510.
52. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. Epub 2013/01/18. doi: 10.1186/1471-2105-14-7. PubMed PMID: 23323831; PMCID: PMC3618321.
53. Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, Nakamura M, Miao S, Cao P, Learned RM, Chen JL, Li Y. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002;277(22):19649-57. Epub 2002/03/06. doi: 10.1074/jbc.M200743200. PubMed PMID: 11877444.
54. Hayes MM, Lane BR, King SR, Markovitz DM, Coffey MJ. Peroxisome proliferator-activated receptor gamma agonists inhibit HIV-1 replication in macrophages by transcriptional and post-transcriptional effects. J Biol Chem. 2002;277(19):16913-9. Epub 2002/02/16. doi: 10.1074/jbc.M200875200. PubMed PMID: 11847231.
55. Joseph SB, Swanstrom R, Kashuba AD, Cohen MS. Bottlenecks in HIV-1 transmission: insights from the study of founder viruses. Nature reviews Microbiology. 2015;13(7):414-25. Epub 2015/06/09. doi: 10.1038/nrmicro3471. PubMed PMID: 26052661; PMCID: PMC4793885.
56. Ortega PAS, Saulle I, Mercurio V, Ibba SV, Lori EM, Fenizia C, Masetti M, Trabattoni D, Caputo SL, Vichi F, Mazzotta F, Clerici M, Biasin M. Interleukin 21 (IL-21)/microRNA-29 (miR-29) axis is associated with natural resistance to HIV-1 infection. AIDS. 2018;32(17):2453-61. Epub 2018/07/14. doi: 10.1097/QAD.0000000000001938. PubMed PMID: 30005016.
57. Ahluwalia JK, Khan SZ, Soni K, Rawat P, Gupta A, Hariharan M, Scaria V, Lalwani M, Pillai B, Mitra D, Brahmachari SK. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology. 2008;5:117. Epub 2008/12/24. doi: 10.1186/1742-4690-5-117. PubMed PMID: 19102781; PMCID: PMC2635386.
58. Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity. 2019;50(5):1132-48. Epub 2019/05/23. doi: 10.1016/j.immuni.2019.04.011. PubMed PMID: 31117010; PMCID: PMC6532429.
59. Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 2009;139(3):499-511. Epub 2009/11/03. doi: 10.1016/j.cell.2009.08.039. PubMed PMID: 19879838; PMCID: PMC2844890.
60. Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451(7177):425-30. Epub 2008/01/18. doi: 10.1038/nature06553. PubMed PMID: 18200009.
61. Dube M, Paquay C, Roy BB, Bego MG, Mercier J, Cohen EA. HIV-1 Vpu antagonizes BST-2 by interfering mainly with the trafficking of newly synthesized BST-2 to the cell surface. Traffic. 2011;12(12):1714-29. Epub 2011/09/10. doi: 10.1111/j.1600-0854.2011.01277.x. PubMed PMID: 21902775; PMCID: PMC3955191.
62. Pardons M, Baxter AE, Massanella M, Pagliuzza A, Fromentin R, Dufour C, Leyre L, Routy JP, Kaufmann DE, Chomont N. Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLoS Pathog. 2019;15(2):e1007619. Epub 2019/02/28. doi: 10.1371/journal.ppat.1007619. PubMed PMID: 30811499; PMCID: PMC6411230.
63. Gosselin A, Wiche Salinas TR, Planas D, Wacleche VS, Zhang Y, Fromentin R, Chomont N, Cohen EA, Shacklett B, Mehraj V, Ghali MP, Routy JP, Ancuta P. HIV persists in CCR6+CD4+ T cells from colon and blood during antiretroviral therapy. AIDS. 2017;31(1):35-48. Epub 2016/11/12. doi: 10.1097/QAD.0000000000001309. PubMed PMID: 27835617; PMCID: PMC5131694.
64. Biancotto A, Grivel JC, Gondois-Rey F, Bettendroffer L, Vigne R, Brown S, Margolis LB, Hirsch I. Dual role of prostratin in inhibition of infection and reactivation of human immunodeficiency virus from latency in primary blood lymphocytes and lymphoid tissue. J Virol. 2004;78(19):10507-15. Epub 2004/09/16. doi: 10.1128/JVI.78.19.10507-10515.2004. PubMed PMID: 15367617; PMCID: PMC516376.
65. Laird GM, Bullen CK, Rosenbloom DI, Martin AR, Hill AL, Durand CM, Siliciano JD, Siliciano RF. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J Clin Invest. 2015;125(5):1901-12. Epub 2015/03/31. doi: 10.1172/JCI80142. PubMed PMID: 25822022; PMCID: PMC4463209.
66. Brooks DG, Hamer DH, Arlen PA, Gao L, Bristol G, Kitchen CM, Berger EA, Zack JA. Molecular characterization, reactivation, and depletion of latent HIV. Immunity. 2003;19(3):413-23. Epub 2003/09/23. doi: 10.1016/s1074-7613(03)00236-x. PubMed PMID: 14499116.
67. Korin YD, Brooks DG, Brown S, Korotzer A, Zack JA. Effects of prostratin on T-cell activation and human immunodeficiency virus latency. J Virol. 2002;76(16):8118-23. Epub 2002/07/23. doi: 10.1128/jvi.76.16.8118-8123.2002. PubMed PMID: 12134017; PMCID: PMC155166.
68. Beans EJ, Fournogerakis D, Gauntlett C, Heumann LV, Kramer R, Marsden MD, Murray D, Chun TW, Zack JA, Wender PA. Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(29):11698-703. Epub 2013/07/03. doi: 10.1073/pnas.1302634110. PubMed PMID: 23812750; PMCID: PMC3718093.
69. Rosen ED, Spiegelman BM. PPARgamma : a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276(41):37731-4. Epub 2001/07/19. doi: 10.1074/jbc.R100034200. PubMed PMID: 11459852.
70. Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, Freiberg AN, Su L, Lee B, Cheng G. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity. 2013;38(1):92-105. Epub 2013/01/01. doi: 10.1016/j.immuni.2012.11.005. PubMed PMID: 23273844; PMCID: PMC3698975.
71. Soroosh P, Wu J, Xue X, Song J, Sutton SW, Sablad M, Yu J, Nelen MI, Liu X, Castro G, Luna R, Crawford S, Banie H, Dandridge RA, Deng X, Bittner A, Kuei C, Tootoonchi M, Rozenkrants N, Herman K, Gao J, Yang XV, Sachen K, Ngo K, Fung-Leung WP, Nguyen S, de Leon-Tabaldo A, Blevitt J, Zhang Y, Cummings MD, Rao T, Mani NS, Liu C, McKinnon M, Milla ME, Fourie AM, Sun S. Oxysterols are agonist ligands of RORgammat and drive Th17 cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(33):12163-8. Epub 2014/08/06. doi: 10.1073/pnas.1322807111. PubMed PMID: 25092323; PMCID: PMC4143045.
72. Santori FR, Huang P, van de Pavert SA, Douglass EF, Jr., Leaver DJ, Haubrich BA, Keber R, Lorbek G, Konijn T, Rosales BN, Rozman D, Horvat S, Rahier A, Mebius RE, Rastinejad F, Nes WD, Littman DR. Identification of natural RORgamma ligands that regulate the development of lymphoid cells. Cell metabolism. 2015;21(2):286-98. Epub 2015/02/05. doi: 10.1016/j.cmet.2015.01.004. PubMed PMID: 25651181; PMCID: PMC4317570.
73. Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M. Induction of the nuclear receptor PPAR-gamma by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol. 2014;15(11):1026-37. Epub 2014/09/30. doi: 10.1038/ni.3005. PubMed PMID: 25263125.
74. Maciolek JA, Pasternak JA, Wilson HL. Metabolism of activated T lymphocytes. Curr Opin Immunol. 2014;27:60-74. Epub 2014/02/22. doi: 10.1016/j.coi.2014.01.006. PubMed PMID: 24556090.
75. Loisel-Meyer S, Swainson L, Craveiro M, Oburoglu L, Mongellaz C, Costa C, Martinez M, Cosset FL, Battini JL, Herzenberg LA, Herzenberg LA, Atkuri KR, Sitbon M, Kinet S, Verhoeyen E, Taylor N. Glut1-mediated glucose transport regulates HIV infection. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(7):2549-54. Epub 2012/02/07. doi: 10.1073/pnas.1121427109. PubMed PMID: 22308487; PMCID: PMC3289356.
76. Hollenbaugh JA, Munger J, Kim B. Metabolite profiles of human immunodeficiency virus infected CD4+ T cells and macrophages using LC-MS/MS analysis. Virology. 2011;415(2):153-9. Epub 2011/05/14. doi: 10.1016/j.virol.2011.04.007. PubMed PMID: 21565377; PMCID: PMC3107887.
77. Valle-Casuso JC, Angin M, Volant S, Passaes C, Monceaux V, Mikhailova A, Bourdic K, Avettand-Fenoel V, Boufassa F, Sitbon M, Lambotte O, Thoulouze MI, Muller-Trutwin M, Chomont N, Saez-Cirion A. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4(+) T Cells and Offers an Opportunity to Tackle Infection. Cell metabolism. 2019;29(3):611-26 e5. Epub 2018/12/26. doi: 10.1016/j.cmet.2018.11.015. PubMed PMID: 30581119.
78. Vignali PDA, Barbi J, Pan F. Metabolic Regulation of T Cell Immunity. Adv Exp Med Biol. 2017;1011:87-130. Epub 2017/09/07. doi: 10.1007/978-94-024-1170-6_2. PubMed PMID: 28875487.
79. Heredia A, Le N, Gartenhaus RB, Sausville E, Medina-Moreno S, Zapata JC, Davis C, Gallo RC, Redfield RR. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(30):9412-7. Epub 2015/07/15. doi: 10.1073/pnas.1511144112. PubMed PMID: 26170311; PMCID: PMC4522811.
80. Besnard E, Hakre S, Kampmann M, Lim HW, Hosmane NN, Martin A, Bassik MC, Verschueren E, Battivelli E, Chan J, Svensson JP, Gramatica A, Conrad RJ, Ott M, Greene WC, Krogan NJ, Siliciano RF, Weissman JS, Verdin E. The mTOR Complex Controls HIV Latency. Cell Host Microbe. 2016;20(6):785-97. Epub 2016/12/16. doi: 10.1016/j.chom.2016.11.001. PubMed PMID: 27978436; PMCID: PMC5354304.
81. Jin S, Liao Q, Chen J, Zhang L, He Q, Zhu H, Zhang X, Xu J. TSC1 and DEPDC5 regulate HIV-1 latency through the mTOR signaling pathway. Emerg Microbes Infect. 2018;7(1):138. Epub 2018/08/09. doi: 10.1038/s41426-018-0139-5. PubMed PMID: 30087333; PMCID: PMC6081400.
82. Hug P, Lin HM, Korte T, Xiao X, Dimitrov DS, Wang JM, Puri A, Blumenthal R. Glycosphingolipids promote entry of a broad range of human immunodeficiency virus type 1 isolates into cell lines expressing CD4, CXCR4, and/or CCR5. J Virol. 2000;74(14):6377-85. Epub 2000/06/23. doi: 10.1128/jvi.74.14.6377-6385.2000. PubMed PMID: 10864648; PMCID: PMC112144.
83. Nguyen DH, Hildreth JE. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol. 2000;74(7):3264-72. Epub 2000/03/09. doi: 10.1128/jvi.74.7.3264-3272.2000. PubMed PMID: 10708443; PMCID: PMC111827.
84. Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(24):13925-30. Epub 2001/11/22. doi: 10.1073/pnas.241320298. PubMed PMID: 11717449; PMCID: PMC61143.
85. Pais-Correia AM, Sachse M, Guadagnini S, Robbiati V, Lasserre R, Gessain A, Gout O, Alcover A, Thoulouze MI. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nature medicine. 2010;16(1):83-9. Epub 2009/12/22. doi: 10.1038/nm.2065. PubMed PMID: 20023636.
86. Thoulouze MI, Alcover A. Can viruses form biofilms? Trends in microbiology. 2011;19(6):257-62. Epub 2011/04/05. doi: 10.1016/j.tim.2011.03.002. PubMed PMID: 21458997.
87. Simon V, Bloch N, Landau NR. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat Immunol. 2015;16(6):546-53. Epub 2015/05/20. doi: 10.1038/ni.3156. PubMed PMID: 25988886; PMCID: PMC6908429.
88. Goujon C, Moncorge O, Bauby H, Doyle T, Ward CC, Schaller T, Hue S, Barclay WS, Schulz R, Malim MH. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature. 2013;502(7472):559-62. Epub 2013/09/21. doi: 10.1038/nature12542. PubMed PMID: 24048477; PMCID: PMC3808269.
89. Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, Bieniasz PD. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature. 2013;502(7472):563-6. Epub 2013/10/15. doi: 10.1038/nature12653. PubMed PMID: 24121441; PMCID: PMC3912734.
90. Shi G, Schwartz O, Compton AA. More than meets the I: the diverse antiviral and cellular functions of interferon-induced transmembrane proteins. Retrovirology. 2017;14(1):53. Epub 2017/11/23. doi: 10.1186/s12977-017-0377-y. PubMed PMID: 29162141; PMCID: PMC5697417.
91. Okumura A, Lu G, Pitha-Rowe I, Pitha PM. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(5):1440-5. Epub 2006/01/26. doi: 10.1073/pnas.0510518103. PubMed PMID: 16434471; PMCID: PMC1360585.
92. Llano M, Kelly T, Vanegas M, Peretz M, Peterson TE, Simari RD, Poeschla EM. Blockade of human immunodeficiency virus type 1 expression by caveolin-1. J Virol. 2002;76(18):9152-64. Epub 2002/08/21. doi: 10.1128/jvi.76.18.9152-9164.2002. PubMed PMID: 12186899; PMCID: PMC136432.
93. Firrito C, Bertelli C, Vanzo T, Chande A, Pizzato M. SERINC5 as a New Restriction Factor for Human Immunodeficiency Virus and Murine Leukemia Virus. Annu Rev Virol. 2018;5(1):323-40. Epub 2018/09/29. doi: 10.1146/annurev-virology-092917-043308. PubMed PMID: 30265629.
94. Barr SD, Smiley JR, Bushman FD. The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 2008;4(2):e1000007. Epub 2008/04/05. doi: 10.1371/journal.ppat.1000007. PubMed PMID: 18389079; PMCID: PMC2279259.
95. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427(6977):848-53. Epub 2004/02/27. doi: 10.1038/nature02343. PubMed PMID: 14985764.
96. Hallenberger S, Bosch V, Angliker H, Shaw E, Klenk HD, Garten W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature. 1992;360(6402):358-61. Epub 1992/11/26. doi: 10.1038/360358a0. PubMed PMID: 1360148.
Supplementary Materials
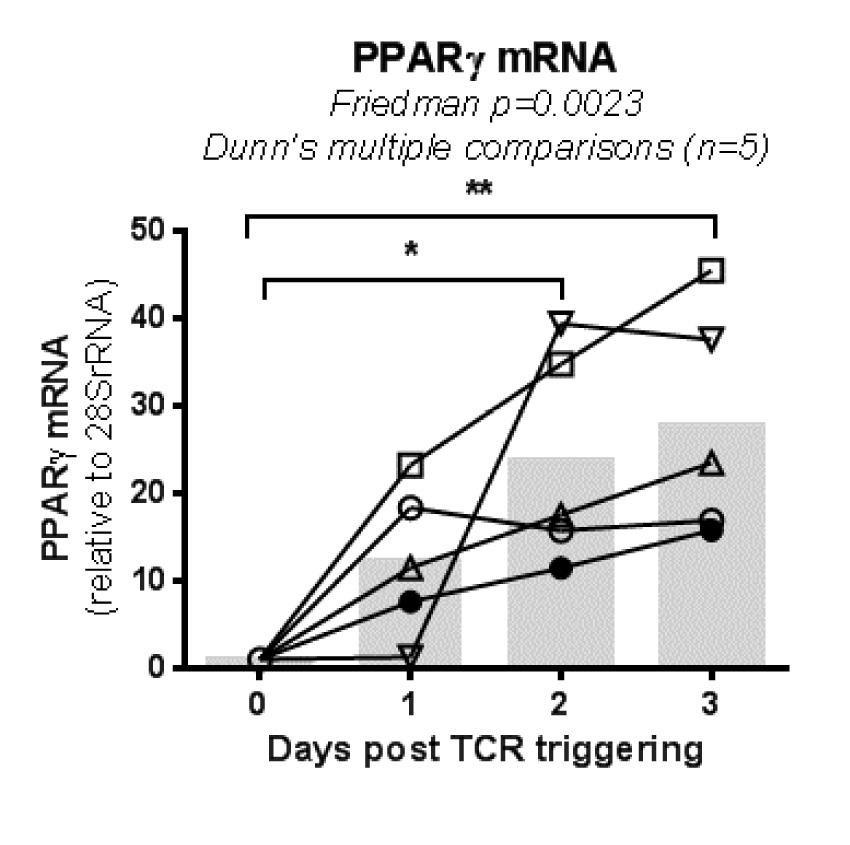
Supplementary Figure 1. PPARy mRNA express is introduced by TCR triggering. RNA was extracted from memory CD4+ T cells from HIV-uninfected individuals (n=5), either freshly-isolated (Day 0) or stimulated CD3/CD28 for 1, 2, or 3 days. PPARy mRNA levels were quantified by RT-PCR (triplicates, 60 ng RNA/test). Each symbol represents results generated with cells from a different individual. Grey bars indicate median PPARy mRNA levels for n=5. Friedman test P-values and Dunn’s multiple comparisons are indicated on the graph.
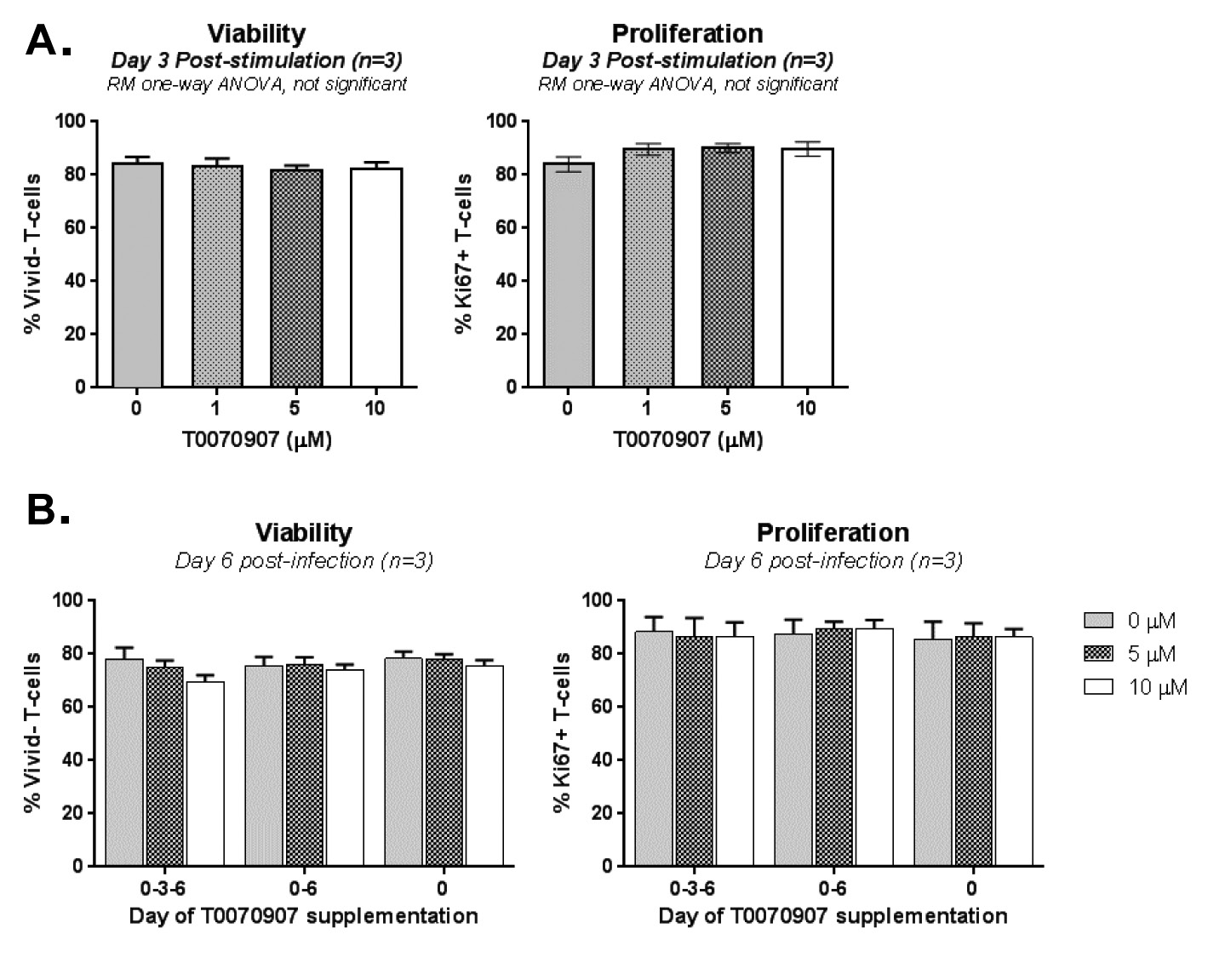
Supplementary Figure 2. Effects of T0070907 of CD4+ T-cell viability and proliferation. (A) Memory CD4+ T cells isolated from HIV-uninfected individuals were 11 stimulated by CD3/CD28 and cultured in the presence/absence of different doses of 12 T0070907 (1, 5, and 10μM) for 3 days, with T0070907 being administered only once. 13 Cells were stained with a viability dye (Vivid), and then intranuclear staining was 14 performed with Ki67 antibodies; Ki67, a marker for cell cycle progression, was used as a 15 surrogate marker of cell proliferation. Shown is cell viability (Vivid- cells, left panel) 16 and proliferation (Ki67+ cells, right panel) measured by flow cytometry with cells from 17 n=3 individuals. (B) Memory CD4+ T cells isolated from HIV-uninfected individuals 18 were stimulated by CD3/CD28, infected with HIVTHRO (as described in Figure 3 legend), 19 and cultured in the presence or the absence of T0070907 (5 and 10 μM). To determine 20 the effect of single versus multiple T0070907 doses on cell viability/proliferation, 21 T0070907 was administered either once at day 0 post-infection (0), twice at days 0 and 6 post-infection (0-6), or every 3 days post-infection (0-3-6). Shown are mean±SD of values obtained with cells from n=3 individuals.
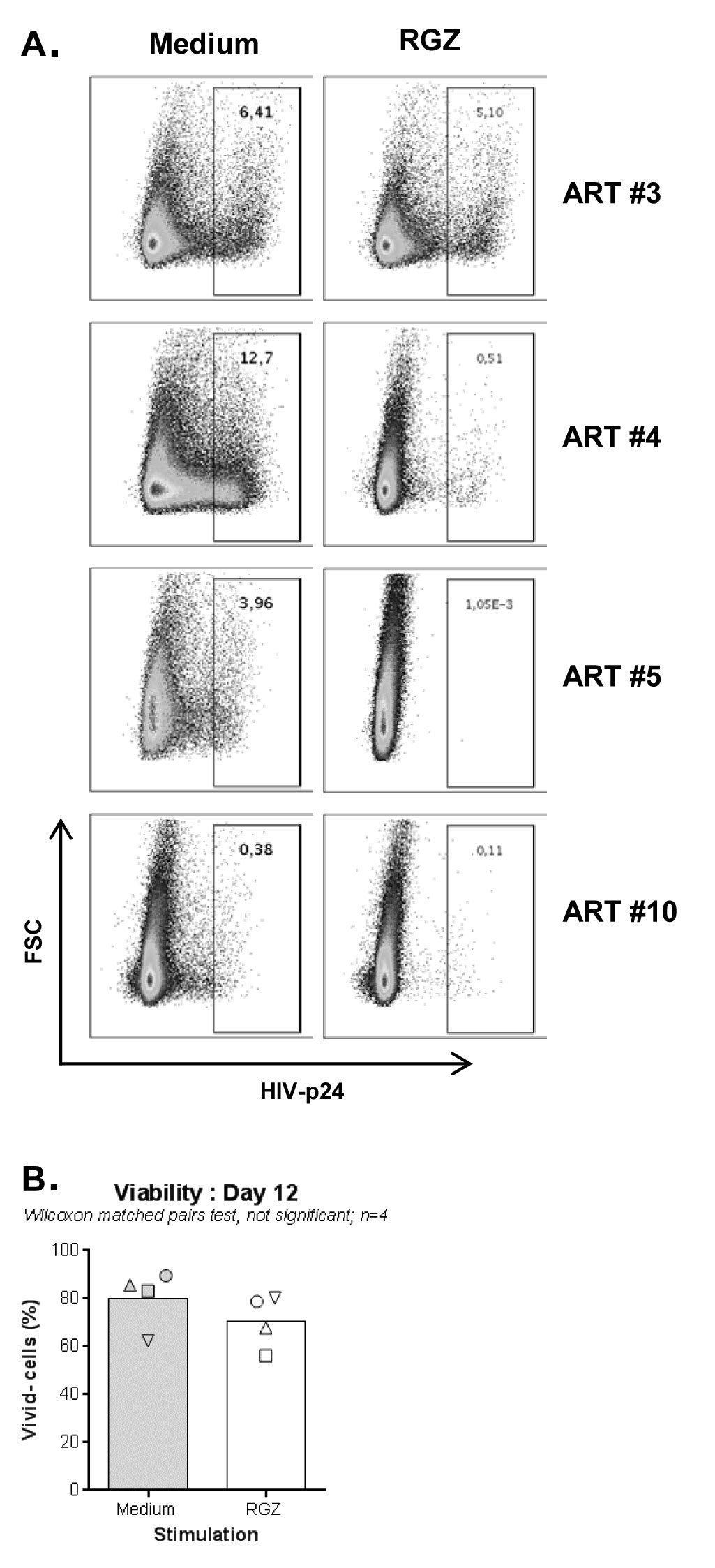
Supplementary Figure 3. RGZ limits HIV outgrowth in memory CD4+ T cells of ART-treated PLWH. A VOA was performed with memory CD4+ T cells of ART treated PLWH (Table 1; ART #3, 4, 5, and 10) in the presence/absence of rosiglitazone (RGZ; 50 µM). At day 12 post-culture, cells were stained as in Figure 2B. Shown is the effect of RGZ on the intracellular expression of HIV-p24 in 4 different ART-treated PLWH (A), as well as the effect of RGZ exposure on cell viability (B). Wilcoxon matched-pairs signed rank test P-values are indicated on the graph in B.
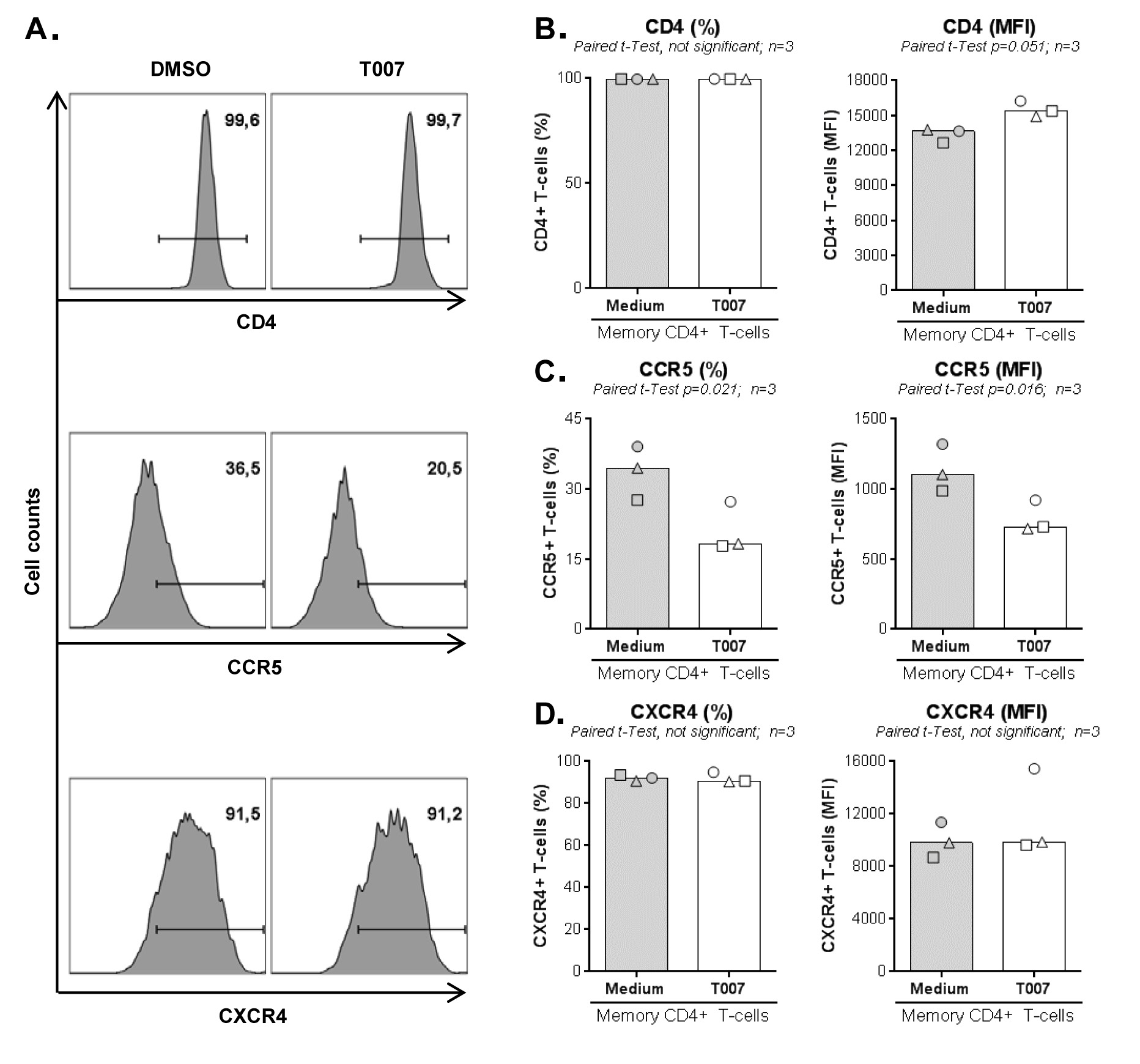
Supplementary Figure 4. T0070907 decreases CCR5 expression on memory CD4+ T cells. Memory CD4+ T cells from HIV-uninfected individuals were stimulated by CD3/CD28 for 3 days and then cultured in the presence/absence of T0070907 (10 µM) for 2 additional days. The expression of HIV-1 receptor CD4 and co-receptors CCR5 and CXCR4 was measured by flow cytometry. Shown are histograms from 1 representative donor (A), and the statistical analyses of CD4 (B), CCR5 (C), and CXCR4 (D) expression(% and MFI) in 3 different donors. Each symbol represents 1 different donor and bars represent median values. Paired t-test values are indicated on the graphs.
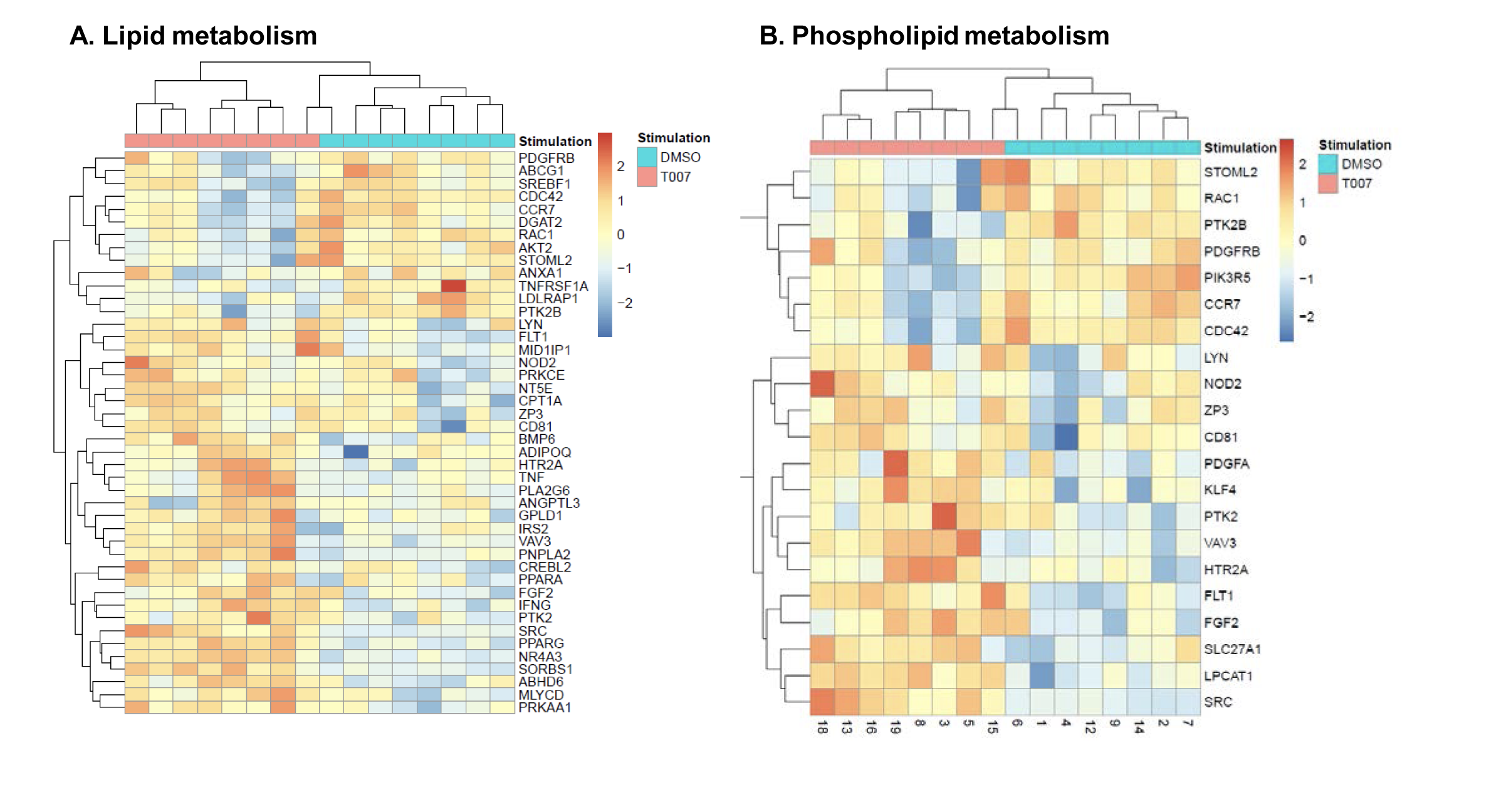
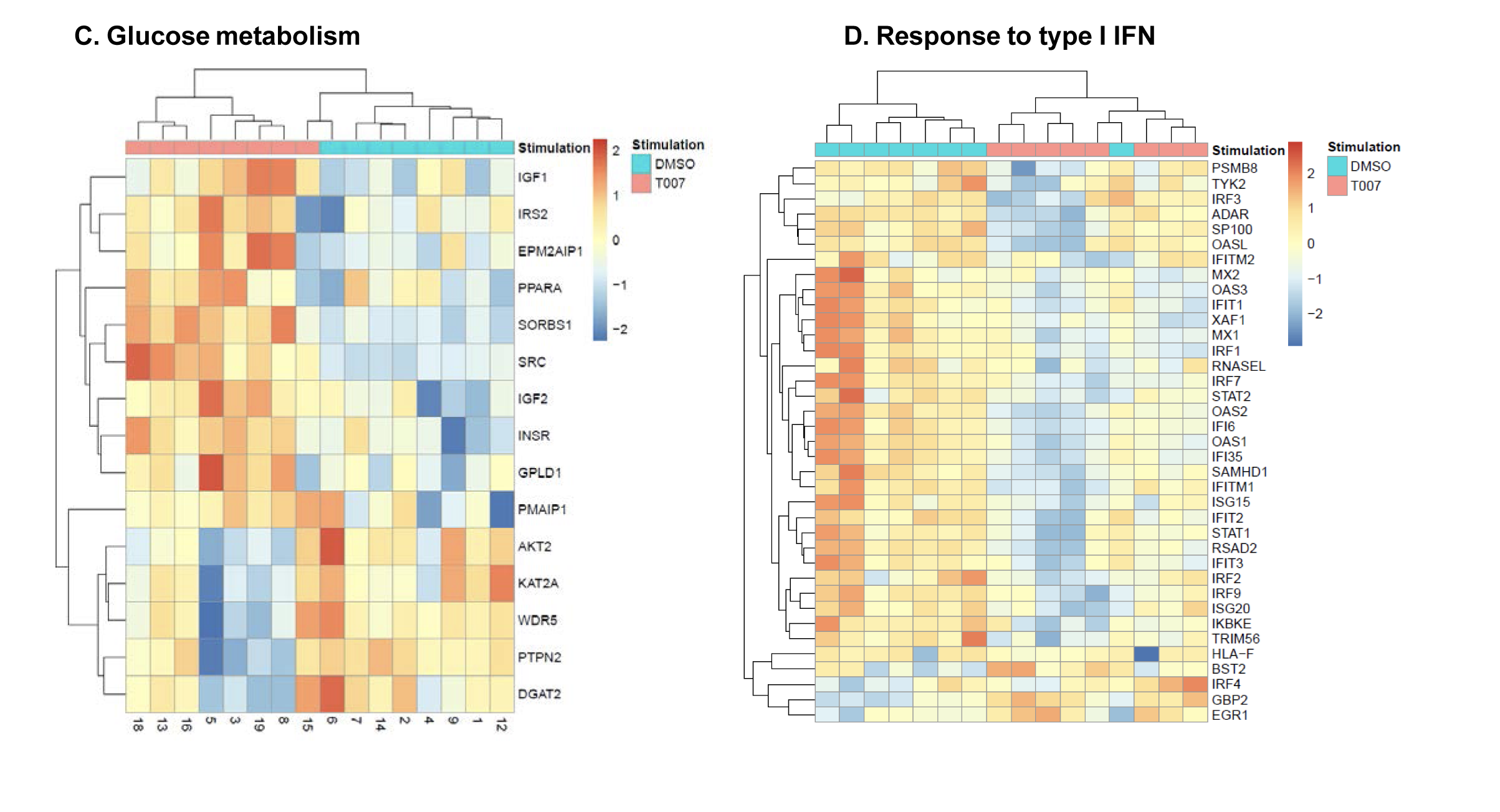
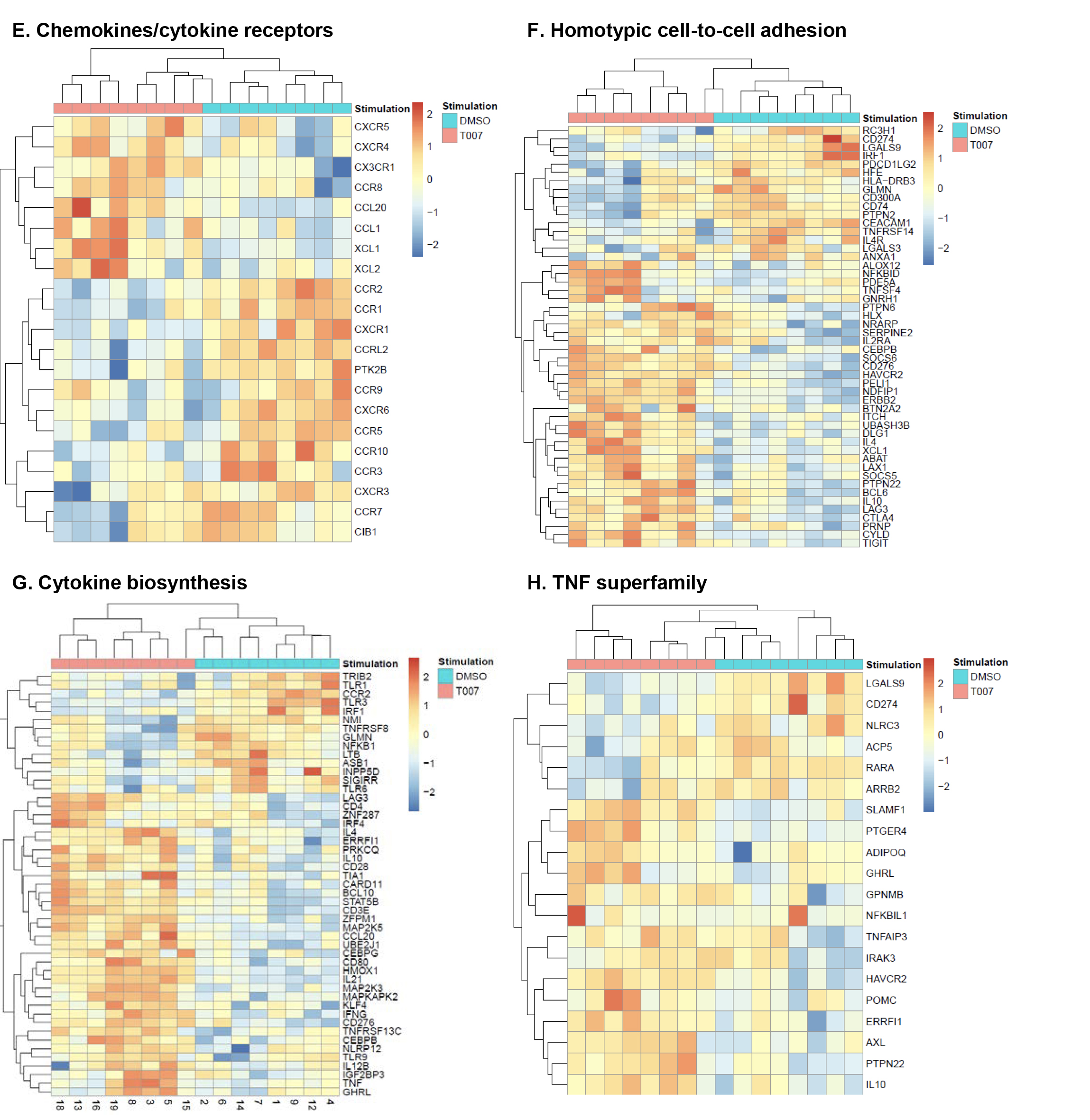
Supplementary Figure 5. Gene Ontology (GO) classification of transcripts modulated by T0070907 in CCR6+ T cells. RNA-Seq transcriptional profiles were generated as described in Figure 5. Differentially expressed genes (P<0.05) were classified based on their biological functions using GO terms as follows: lipid metabolism (A), phospholipid metabolism (B), glucose metabolism (C), response to type I IFN (D), chemokines/cytokines receptors (E), homotypic cell-to-cell adhesion (F), cytokine biosynthesis (G), and TNF superfamily (H). Heatmap cells are scaled by the expression level z-scores for each probe individually. For each heatmap, gene expression values are represented as a gradient from red (highest expression) to blue (lowest expression). Results were generated with cells from n=8 donors, with each column representing 1 donor.
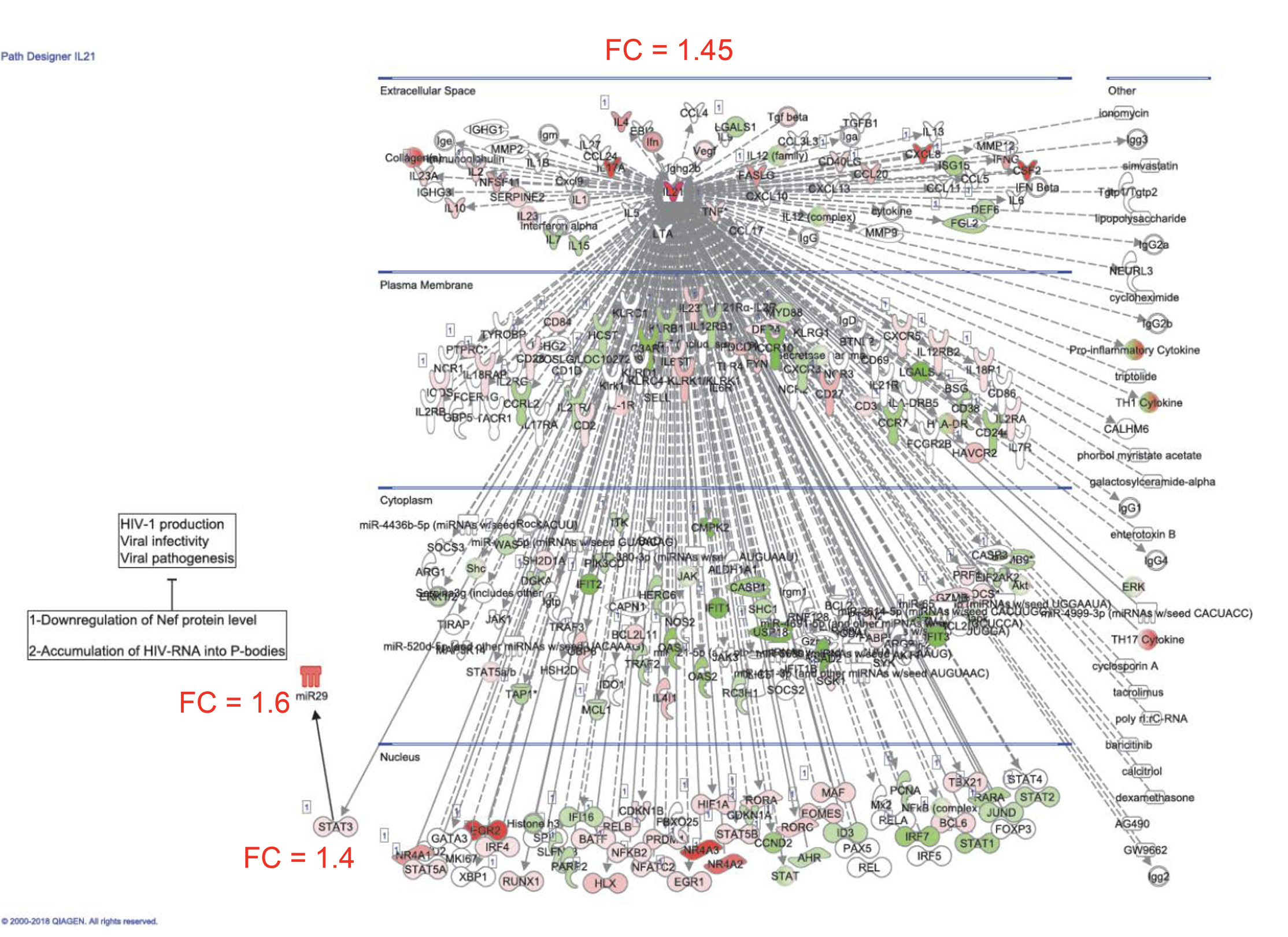
Supplementary Figure 6. Components of the IL-21 signalizing pathway modulated by T0070907 in CCR6+ T cells. RNA-Seq transcriptional profiles were generated as described in Figure 5. Ingenuity pathway analysis (IPA) was used to illustrate gene networks associated with IL-21. The color code is based on the expression FC in T0070907-treated CCR6+ versus untreated CCR6+ T cells (red and green for upregulated and downregulated transcripts, respectively). FC expression relative to IL-21, STAT3, and miR-29 in T0070907-treated versus untreated CCR6+ T cells are illustrated. The miR-29 connection to the IL-21 network was added manually based on a recent publication demonstrating that IL-21 regulates its expression by STAT3 (75).
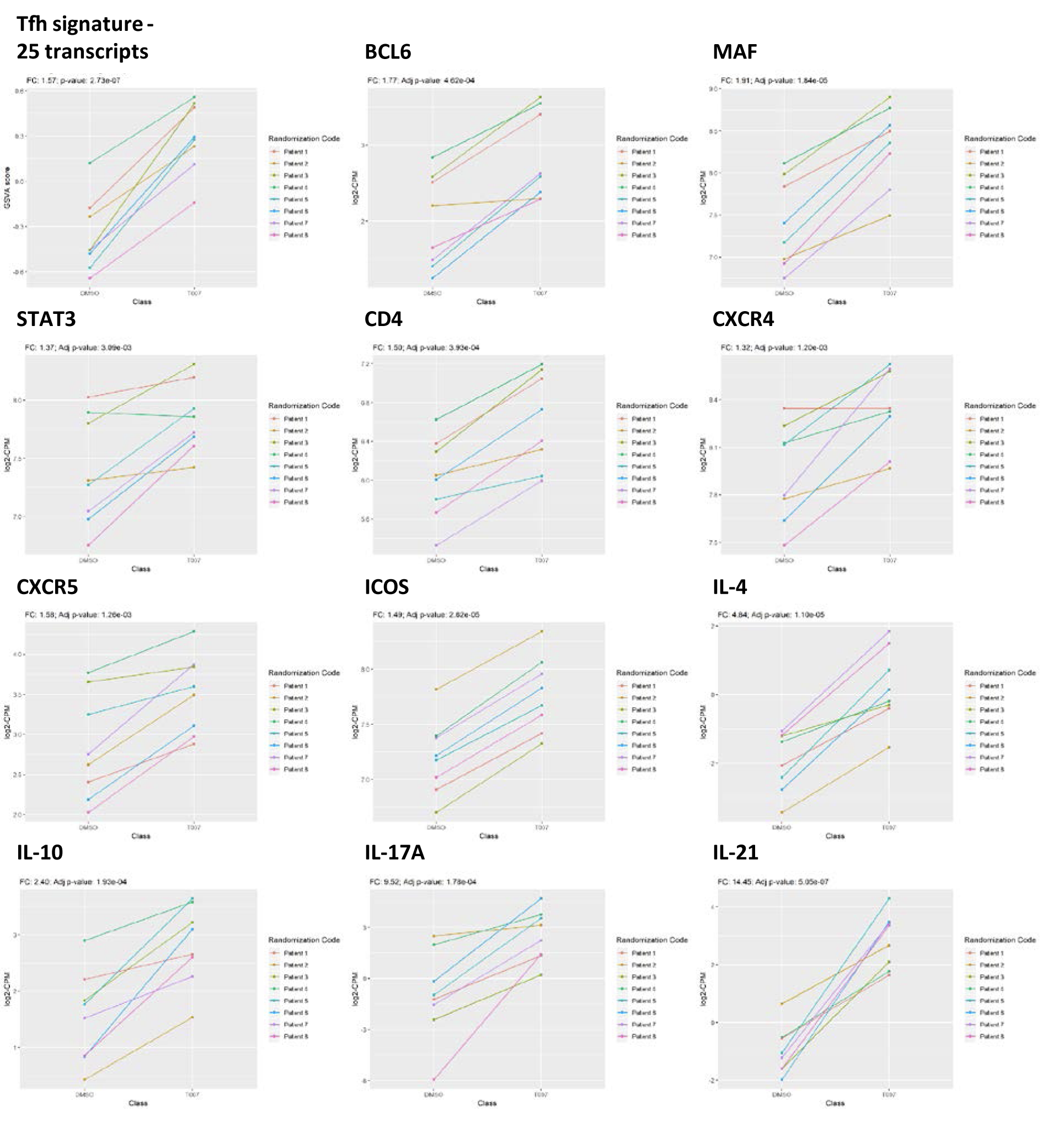
Supplementary Figure 7. T0070907 acts on CCR6+ T cells to promote expression of follicular helper T-cell (Tfh) marker transcripts. RNA-Seq transcriptional profiles were generated as described in Figure 5. An intelligent search was performed to identify Tfh-specific transcripts among transcripts upregulated by T0070907 in CCR6+ T cells. Shown are changes in the expression of 25 Tfh-specific transcripts identified in the literature (Crotty, 2019), as well as individual Tfh-specific transcription factors (BCL6, MAF, and STAT3), surface molecules (CD4, CXCR4, CXCR5, and ICOS), and cytokines (IL-4, IL-10, IL-17A, and IL-21). Results from n=8 individual donors are represented in different colors.
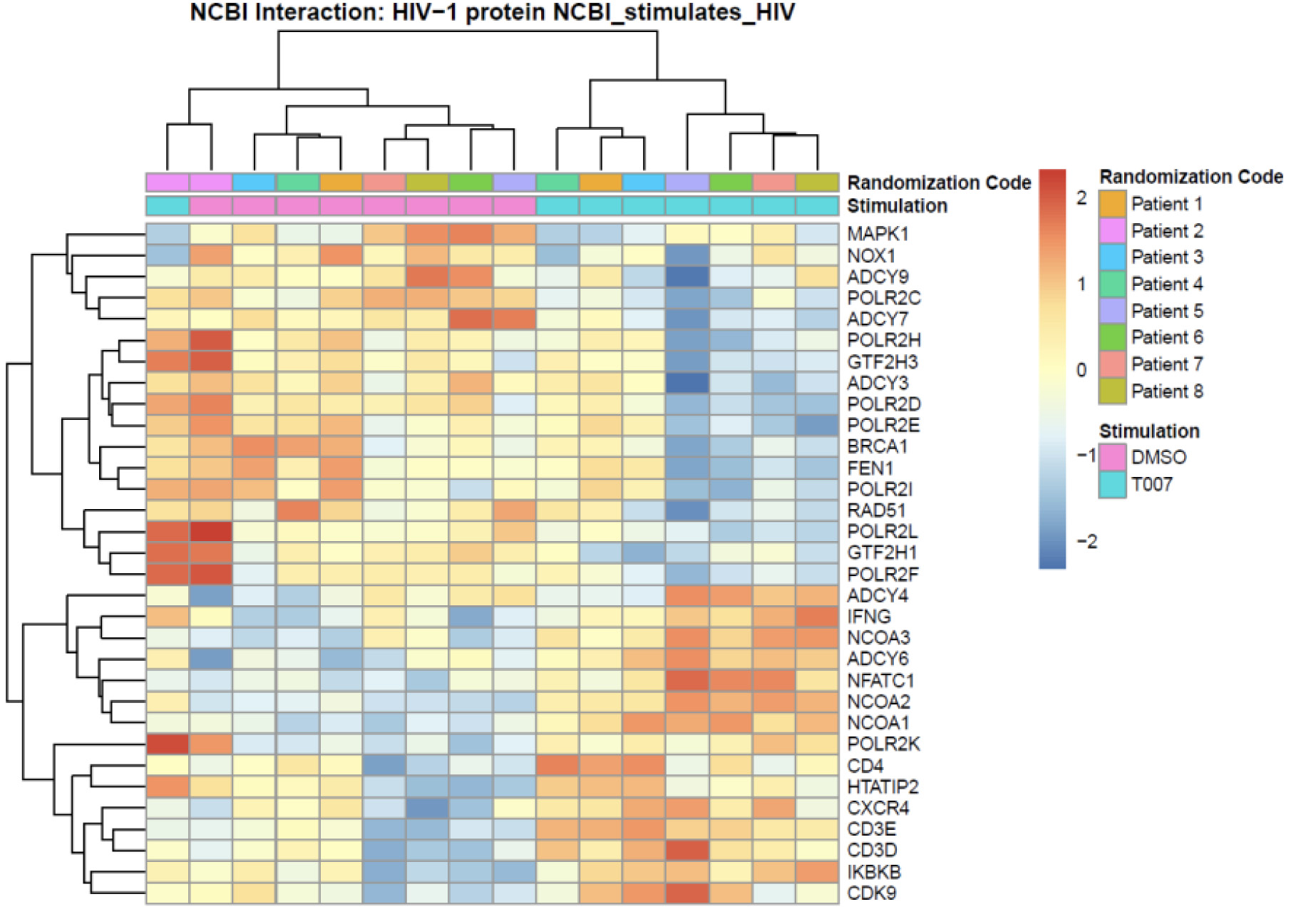
Supplementary Figure 8. Meta-analysis using the NCBI HIV interaction database. RNA-Seq transcriptional profiles were generated as described in Figure 5. Transcripts differentially expressed in CCR6+ T cells cultured in the presence/absence of T0070907 (P< 0.05, FC cut-off 1.3) were matched to the lists of human genes previously identified to interact with HIV-1 proteins (NCBI Interactor database). Heatmap cells are scaled by the expression level z-scores for each probe individually. For each heatmap, gene expression values are represented as a gradient from red (highest expression) to blue (lowest expression). Results from each donor are indicated with a different color code (n=8).
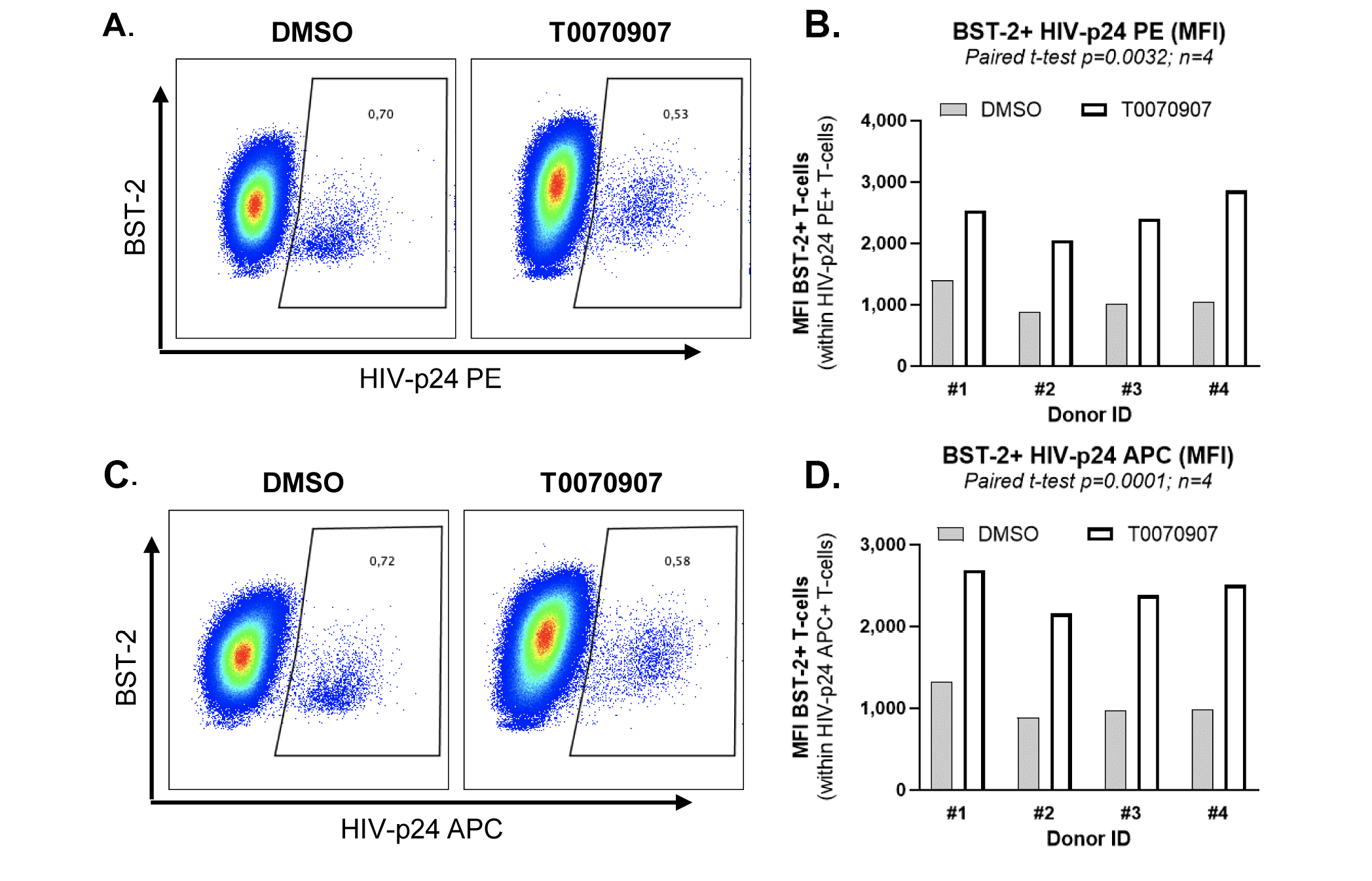
Supplementary Figure 9. T0070907 prevents BST-2 downregulation on HIV-infected cells. Experiments were performed as described in Figure 7 legend. Shown are dot plots of BST-2 and HIVp-24 PE or HIV-p24 APC co-expression (A and C), as well as the statistical analyses for BST-2 expression (MFI) on HIV-p24 PE+ and HIV-p24 APC+ T different donors (B and D). Paired t-test values are indicated on the graphs.
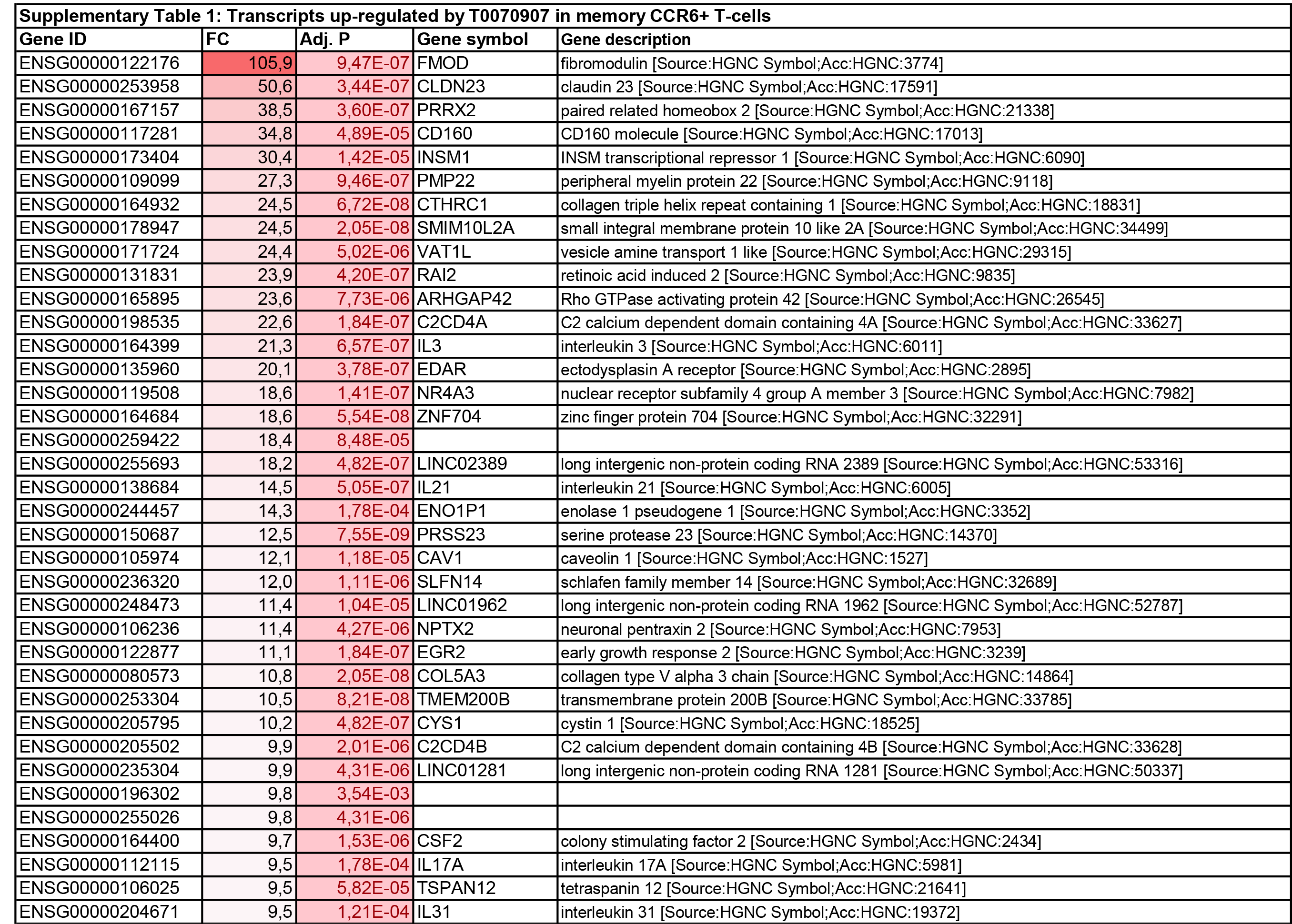
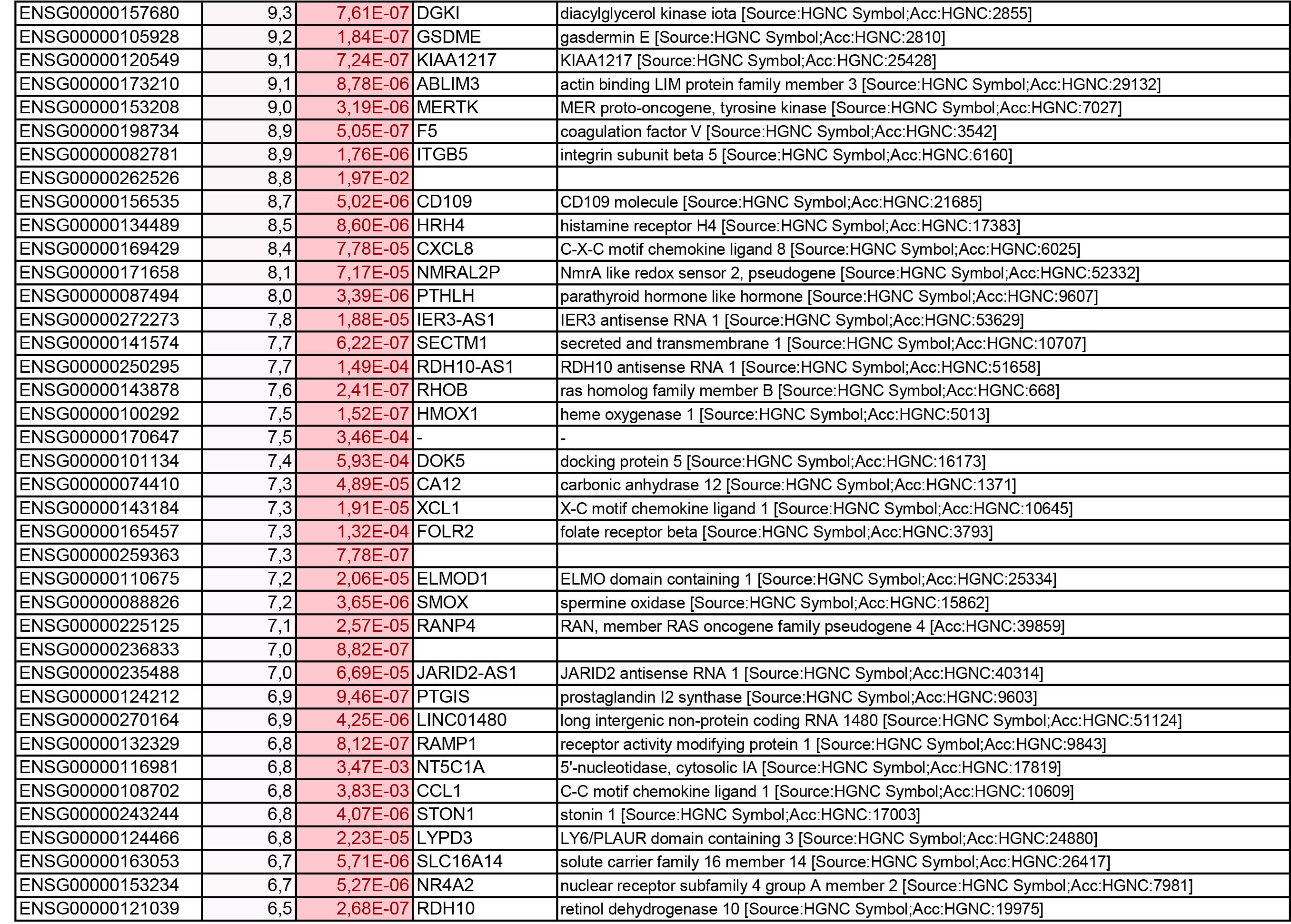

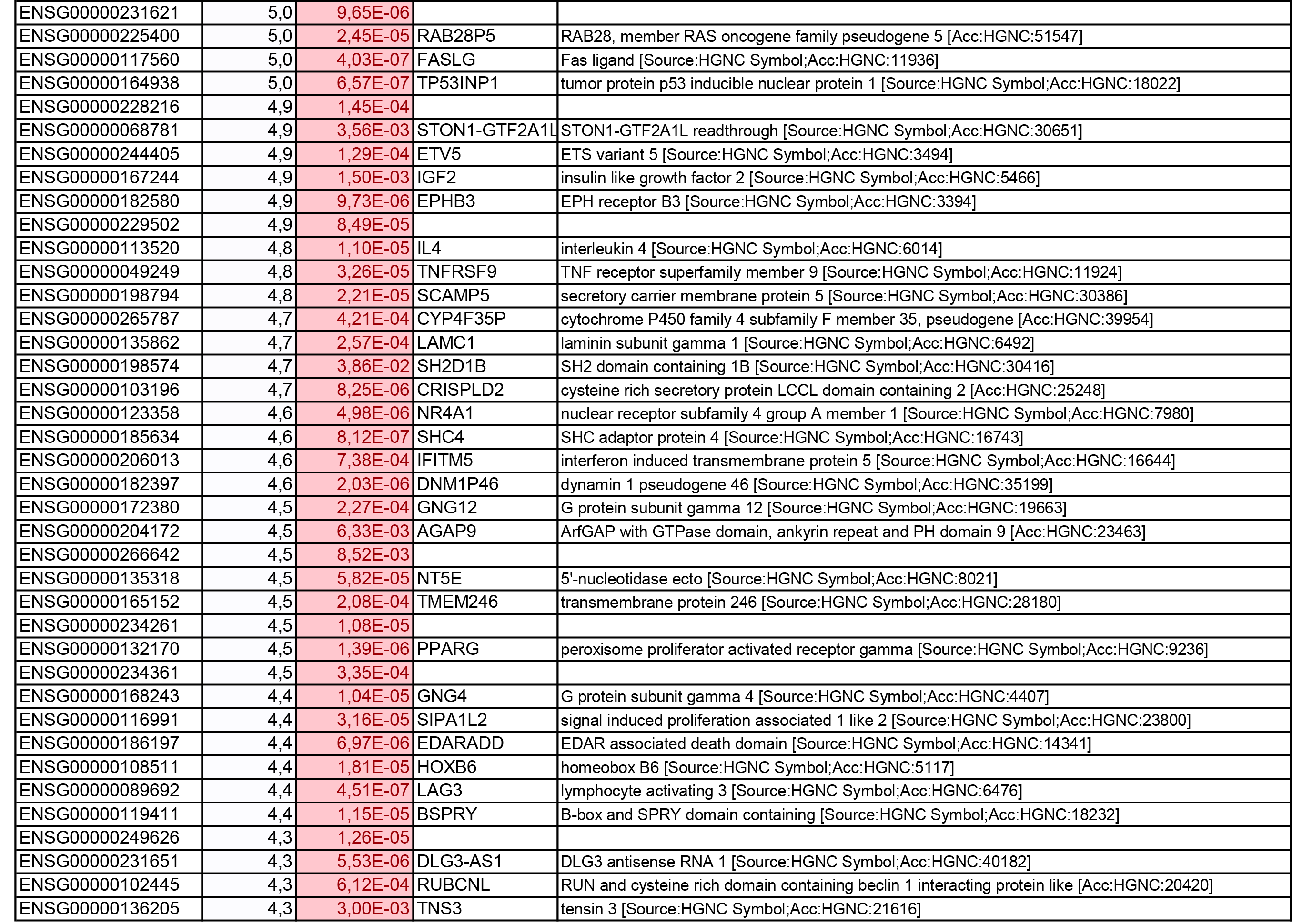
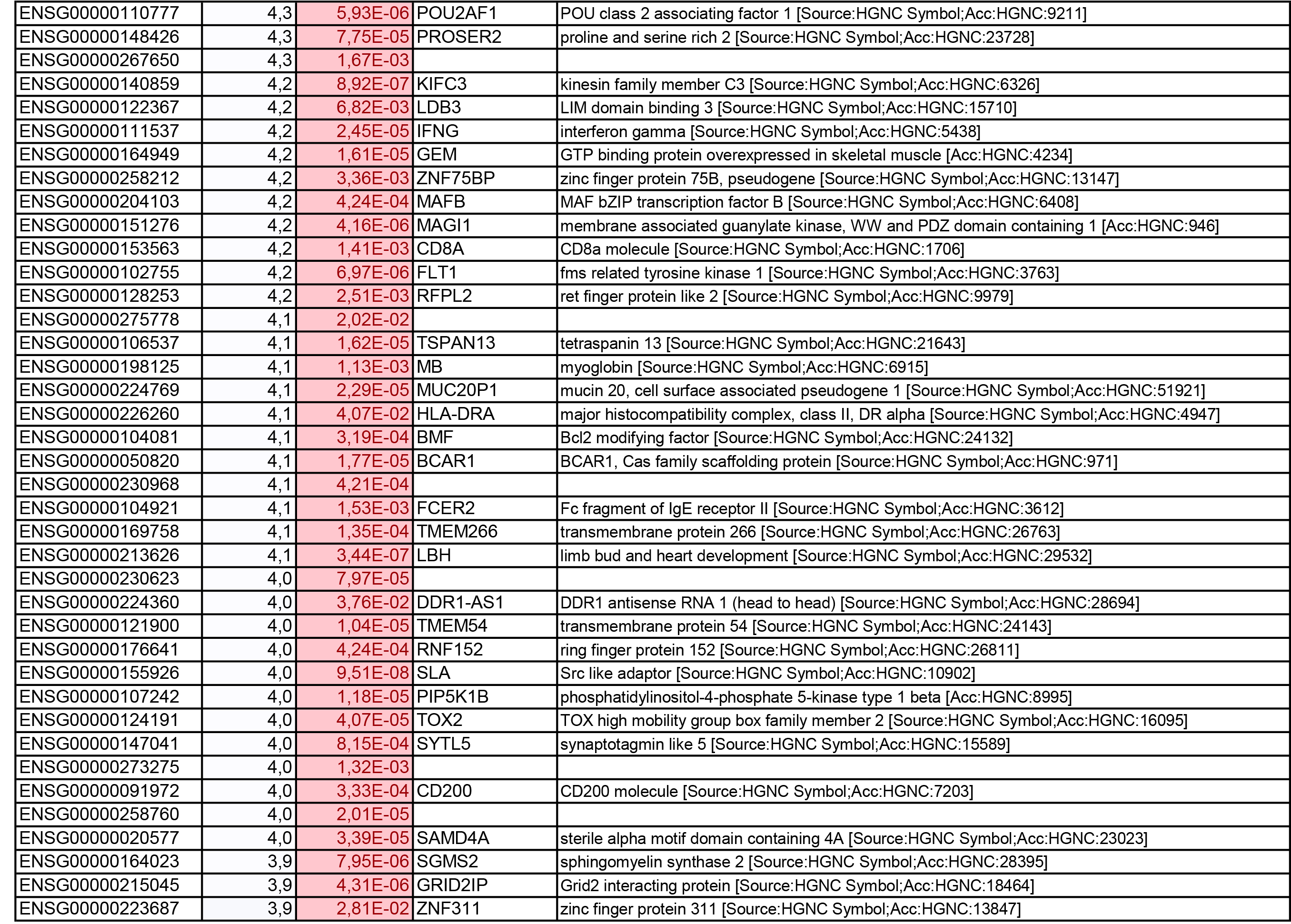
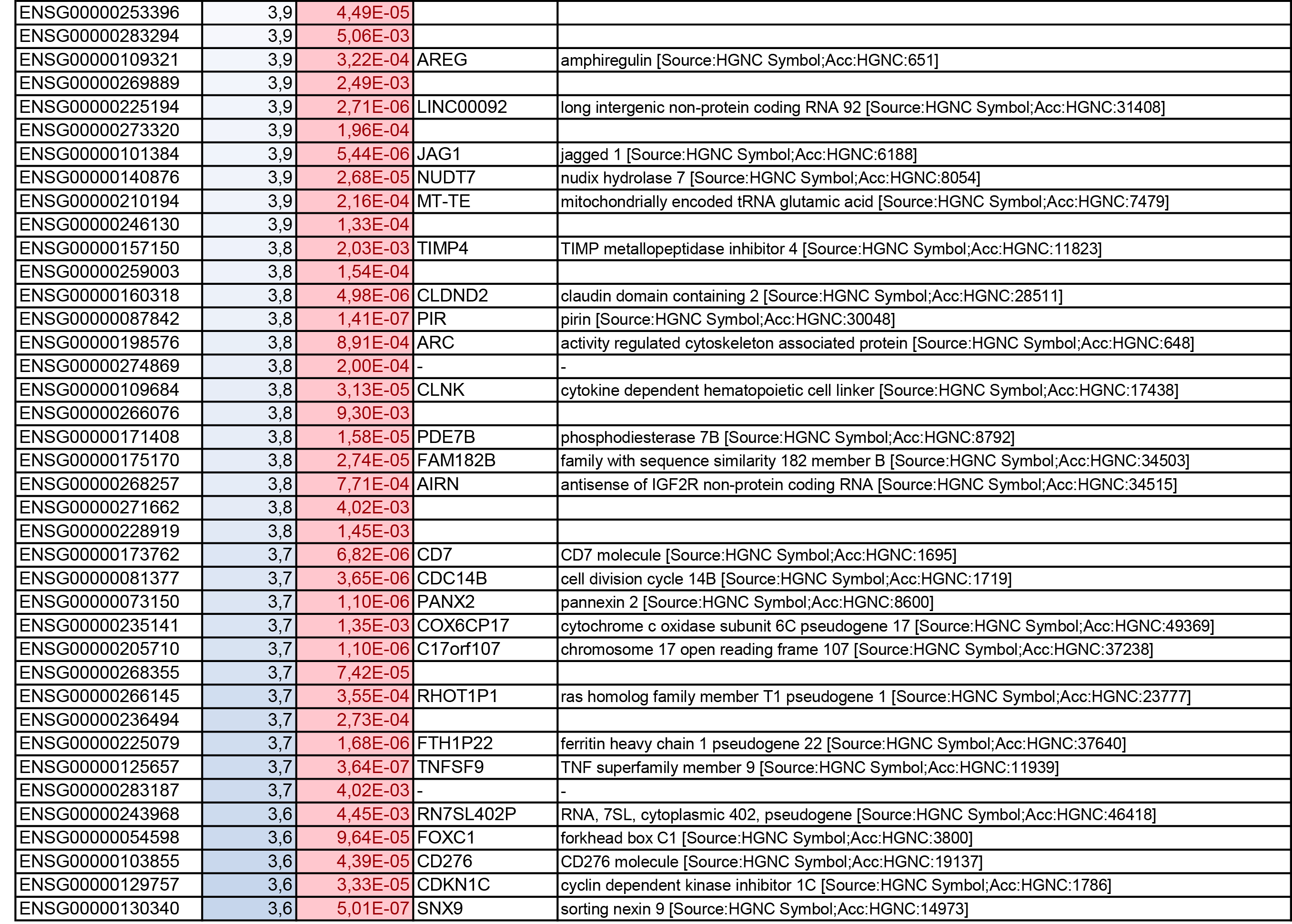
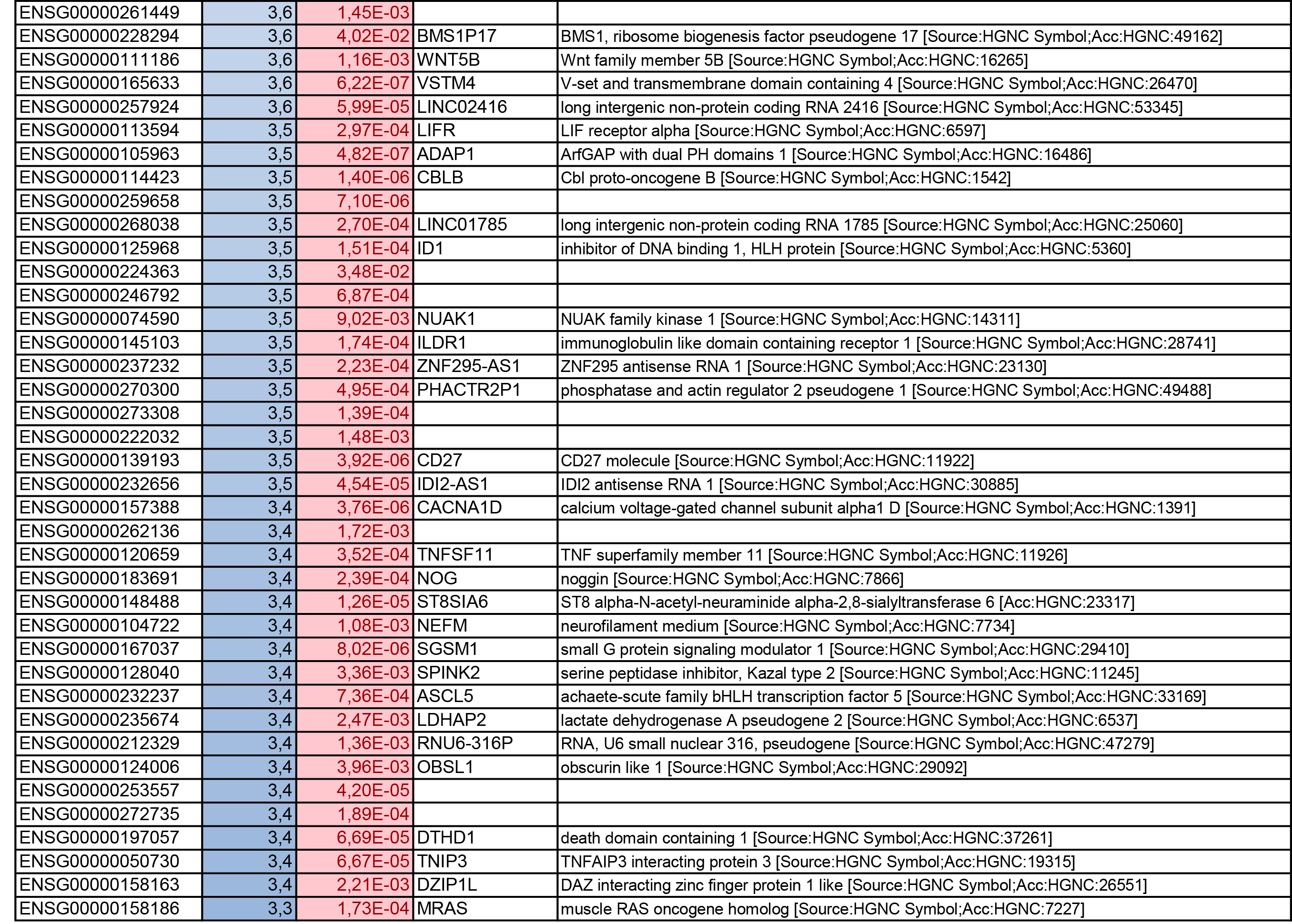
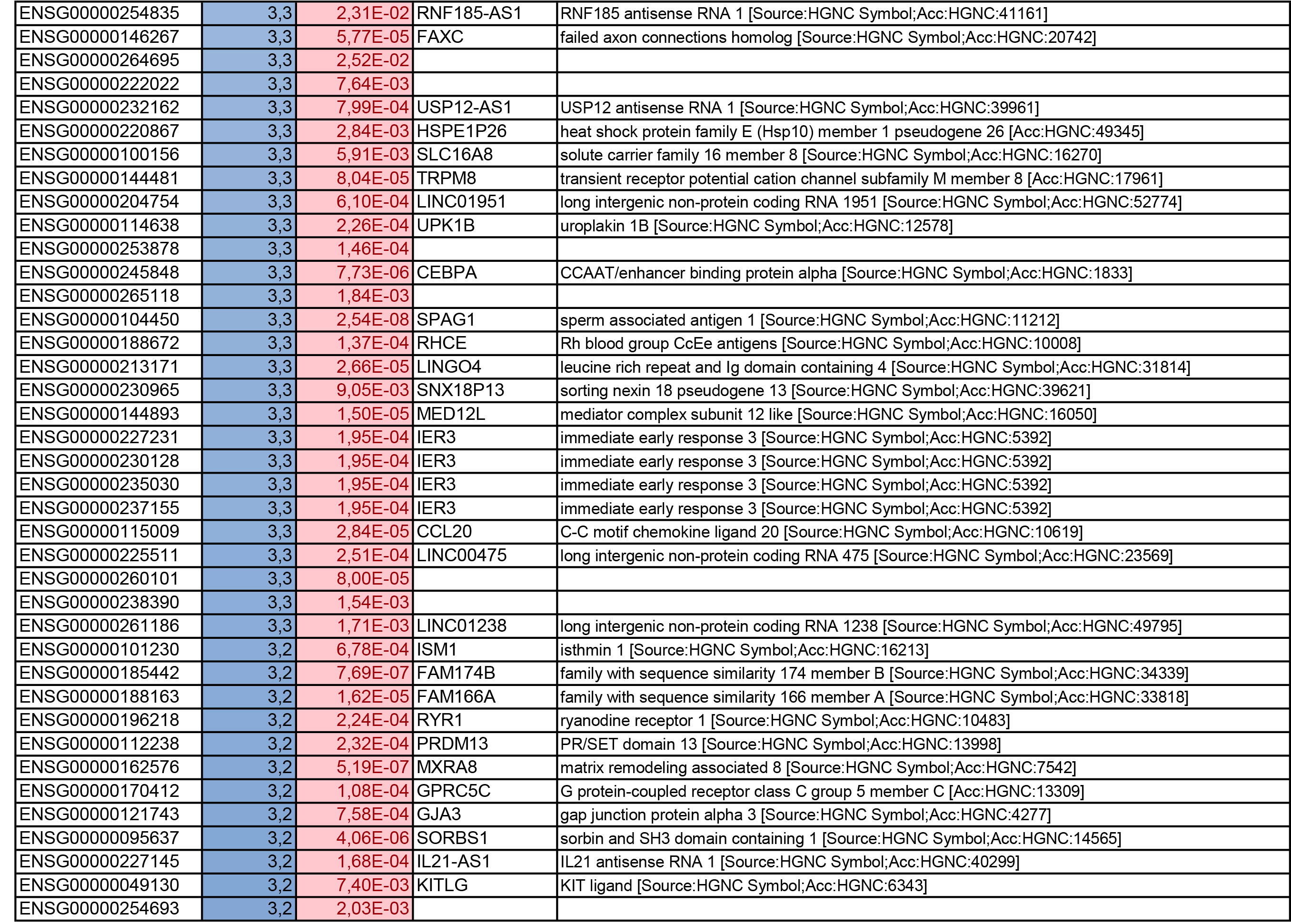
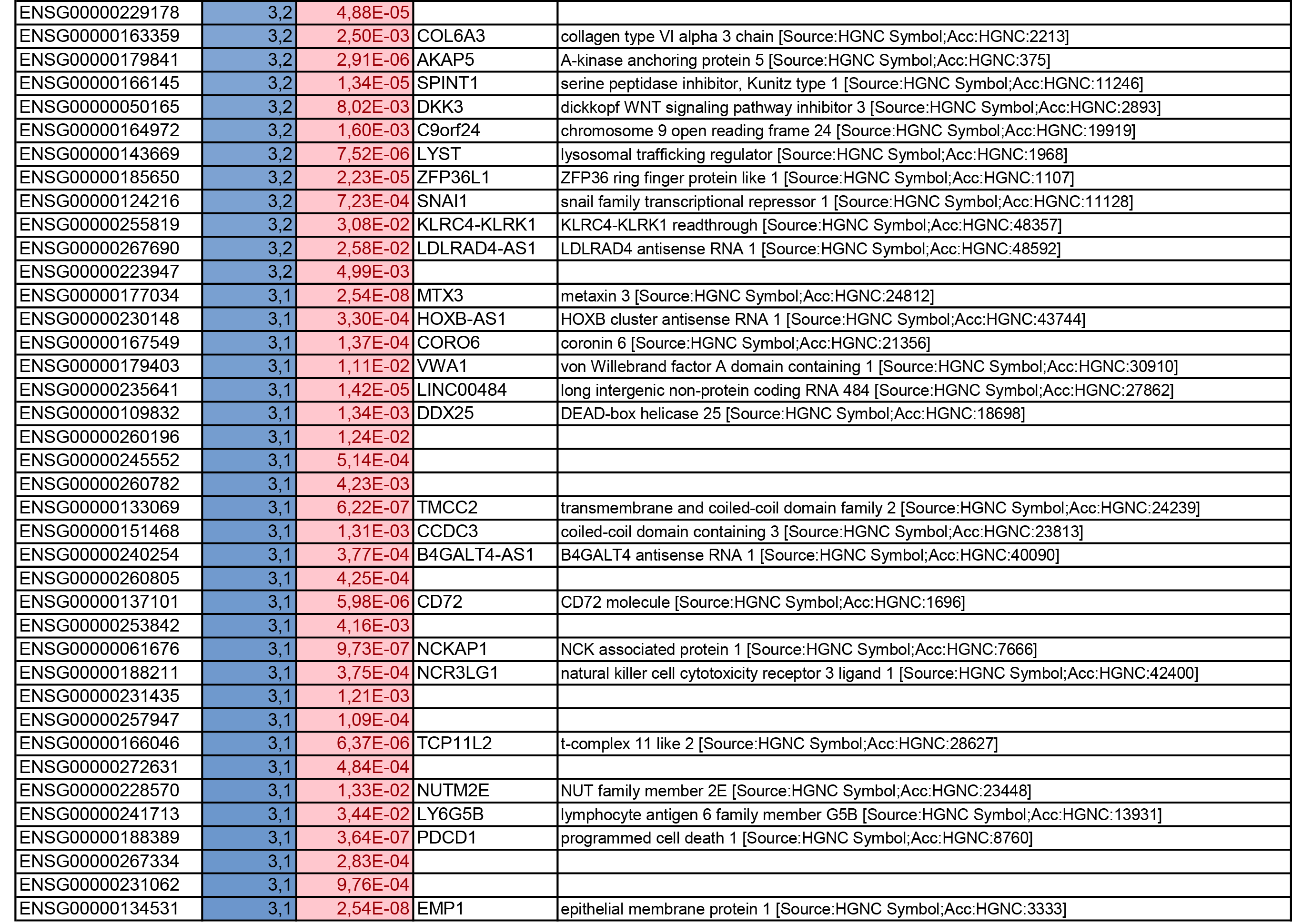
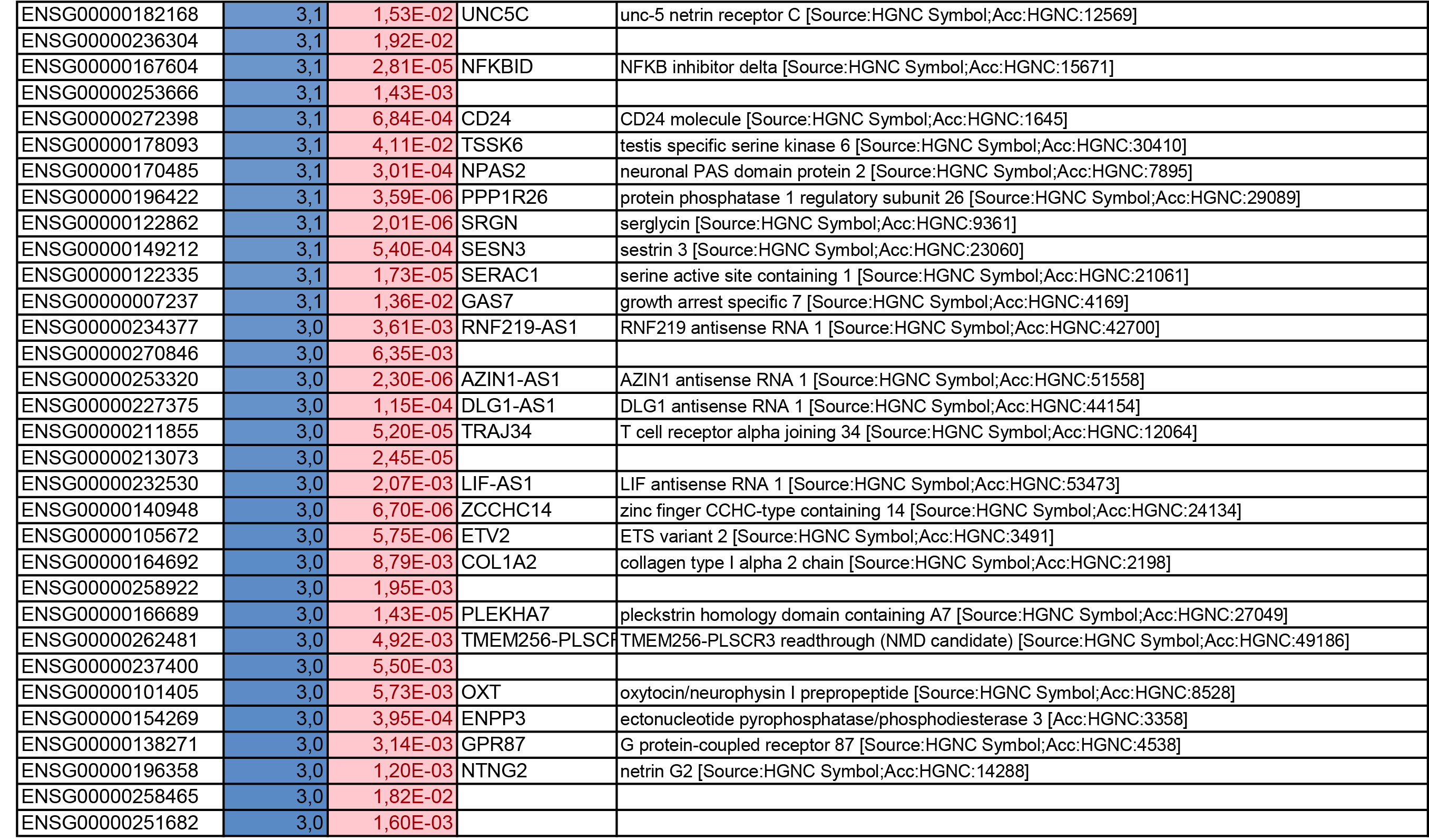
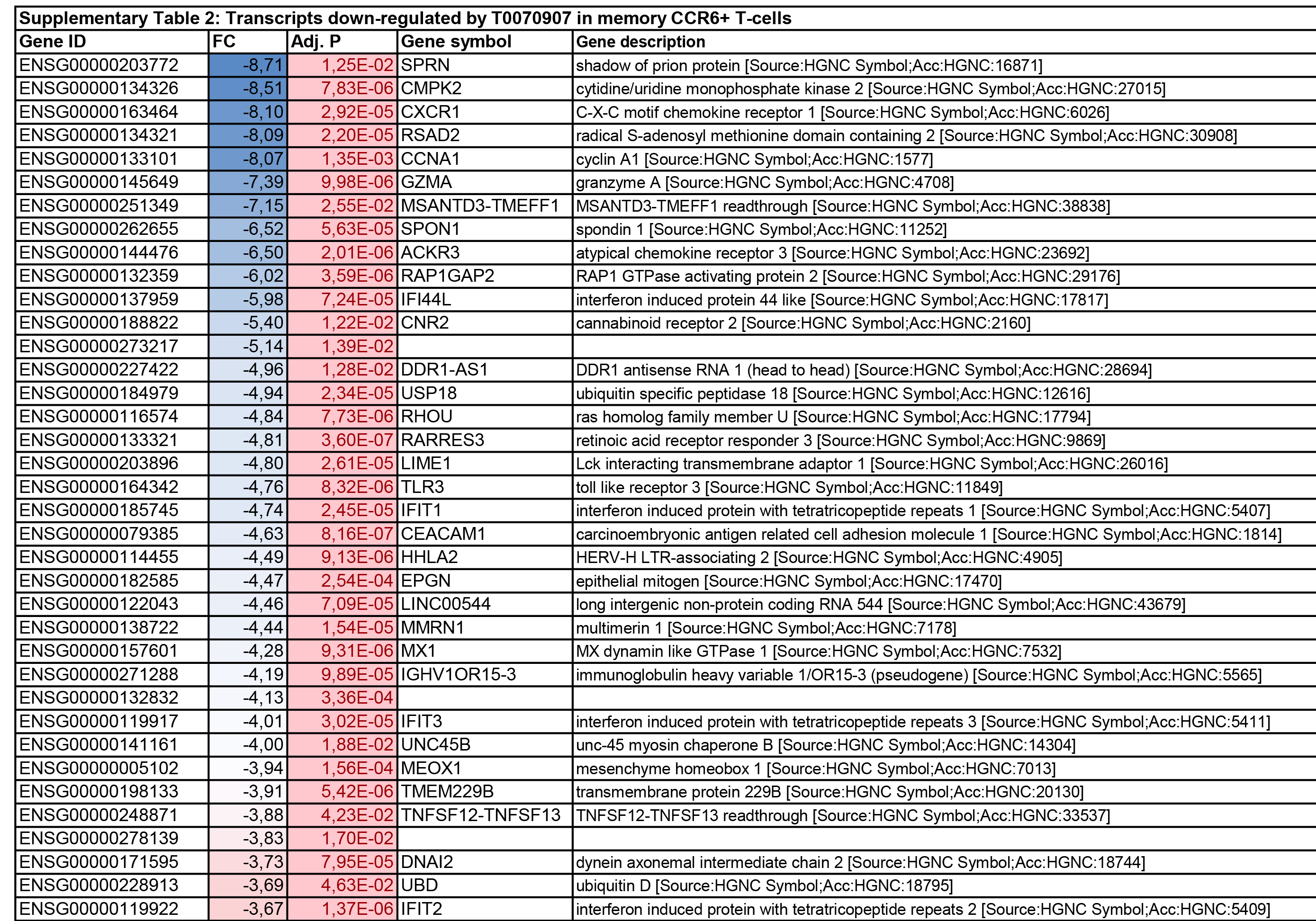
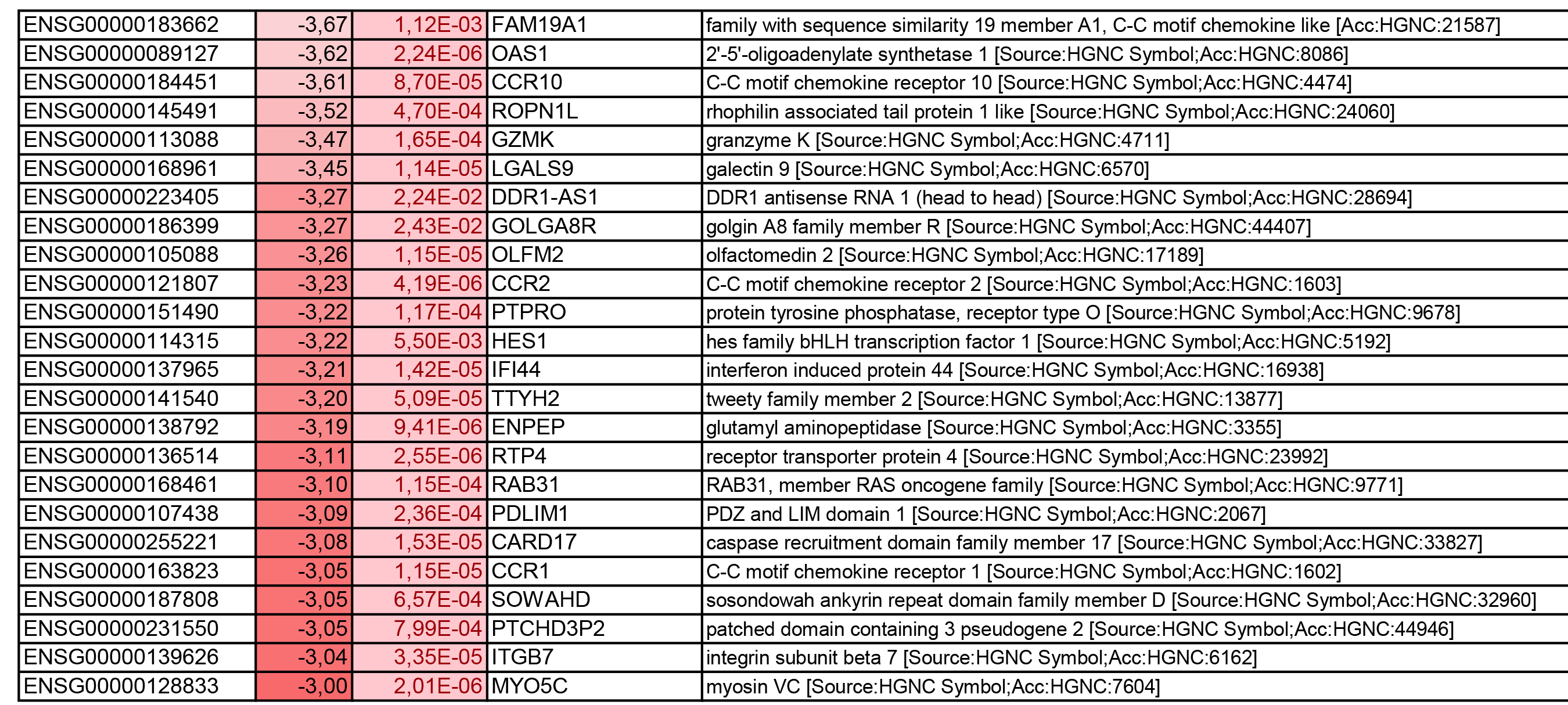
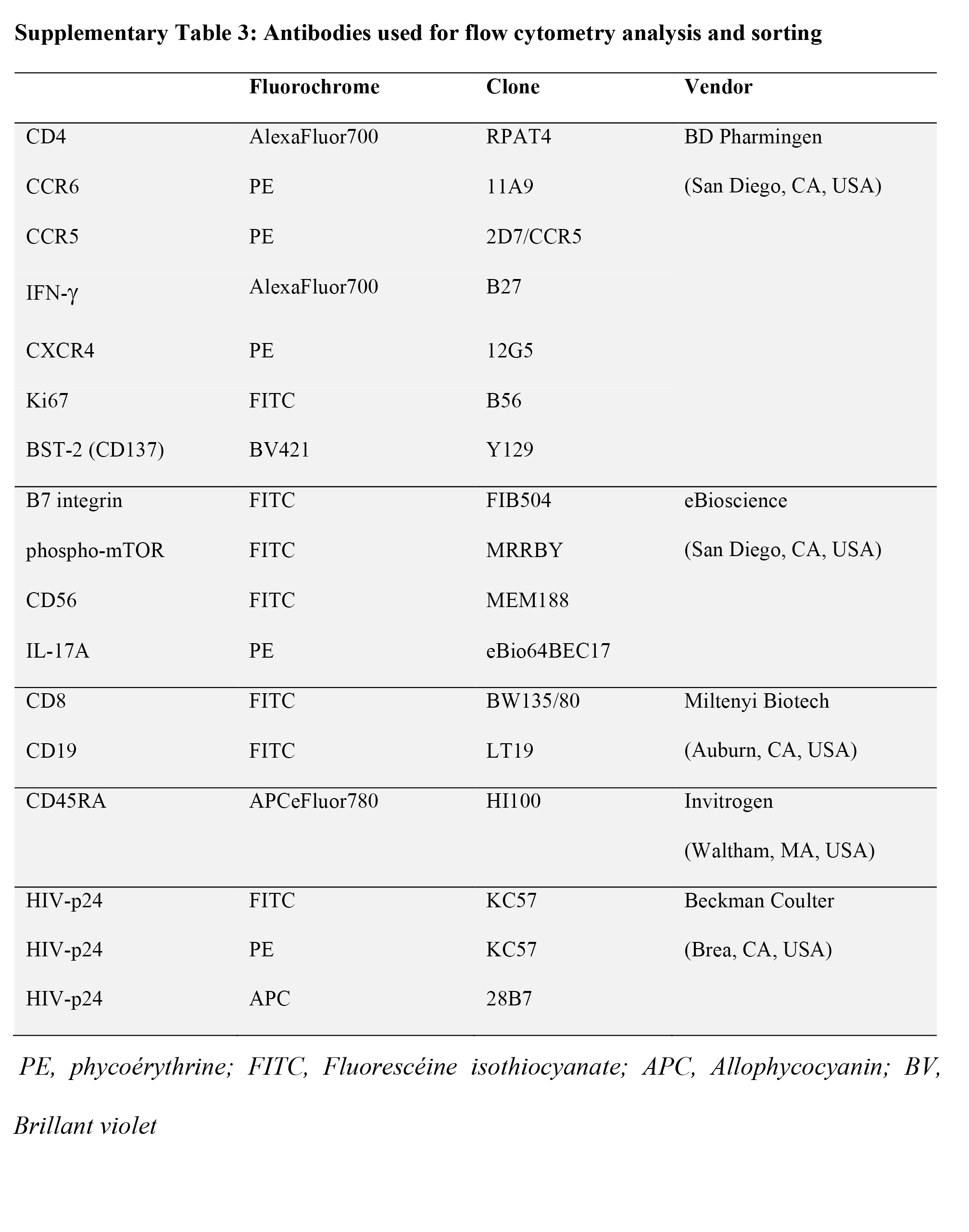

Submitted January 8, 2020 | Accepted April 4, 2020 | Published September 12, 2020