Neetu M. Gulati,1,2* Masaru Miyagi,1 Mayim E. Wiens,3 Jason G. Smith,3 Phoebe L. Stewart1,2
1
Department of Pharmacology, Case Western Reserve University, Cleveland,
Ohio
2
Cleveland Center for Membrane and Structural Biology, Case Western Reserve
University, Cleveland, Ohio
3
Department of Microbiology, University of Washington, Seattle, Washington
Jason G. Smith
jgsmith2@uw.edu
206-685-6144
Phoebe L. Stewart
pls47@case.edu
216-368-4349
*Present address: Neetu M. Gulati, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, USA
Background: Human papillomavirus (HPV) is linked to nearly all cases of cervical cancer. Despite available vaccines, a deeper understanding of the immune response to HPV is needed. Human α-defensin 5 (HD5), an innate immune effector peptide, blocks infection of multiple serotypes of HPV, including high-risk HPV16. While a common mechanism of α-defensin anti-viral activity against nonenveloped viruses such as HPV has emerged, there is limited understanding of how α-defensins bind to viral capsids to block infection.
Methods: We have used cryo-electron microscopy (cryoEM), mass spectrometry (MS) crosslinking and differential lysine modification studies, and molecular dynamics (MD) simulations to probe the interaction of HPV16 pseudovirions (PsVs) with HD5.
Results: CryoEM single particle reconstruction did not reveal HD5 density on the capsid surface. Rather, increased density was observed under the capsid shell in the presence of HD5. MS studies indicate that HD5 binds near the L1 and L2 capsid proteins and specifically near the C-terminal region of L1. MD simulations indicate that favorable electrostatic interactions can be formed between HD5 and the L1 C-terminal tail.
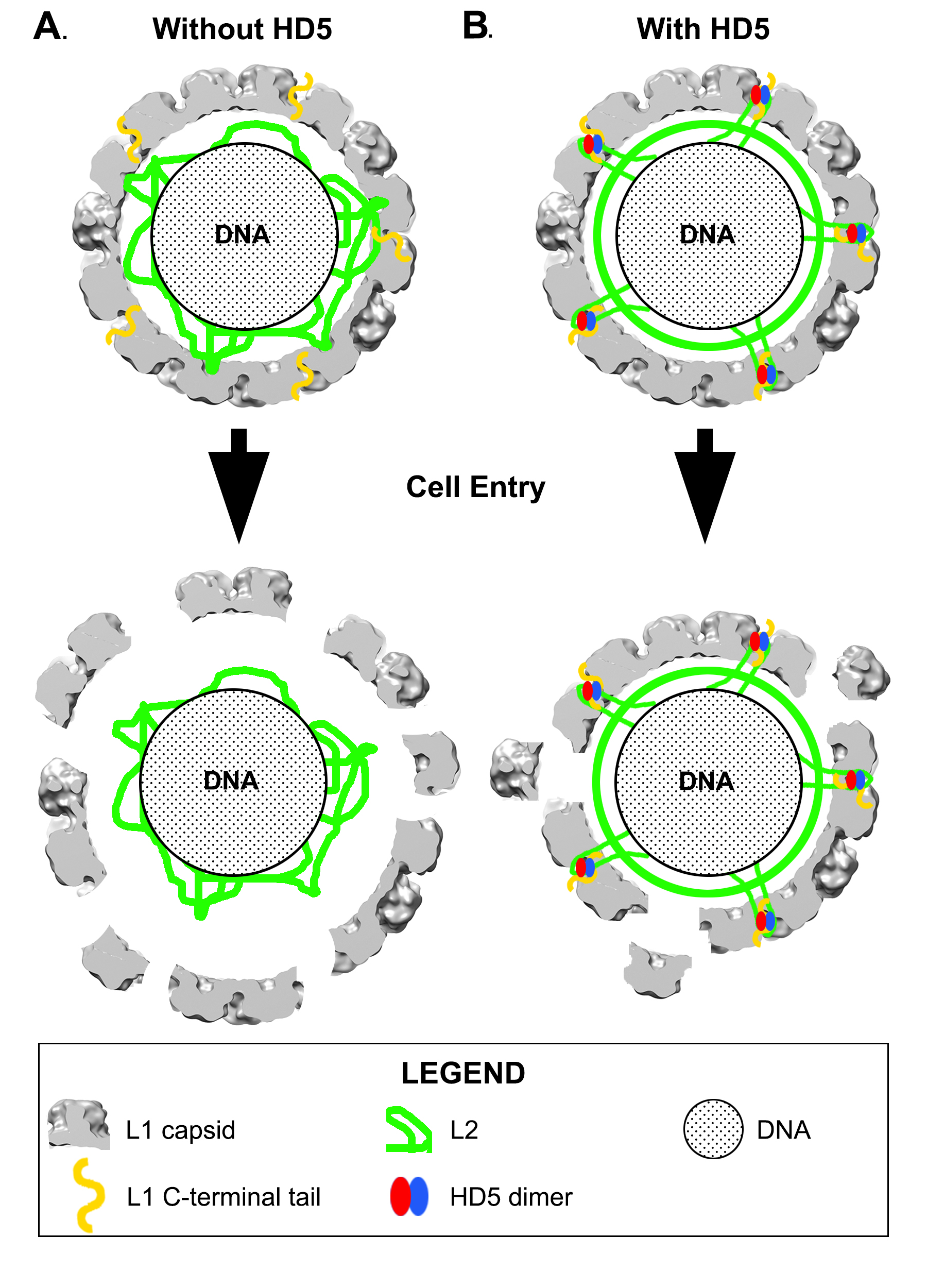
Conclusions: A model is presented for how HD5 affects HPV16 structure and cell entry. In this model, HD5 binds to disordered regions of L1 and L2 protruding from the icosahedrally ordered capsid. HD5 acts to cement interactions between L1 and L2 and leads to a closer association of the L2/genome core with the L1 capsid. This model provides a structural rationale for our prior observation that HD5 interferes with the separation of L1 from the L2/genome complex during cell entry.
Keywords: alpha-Defensins; Human papillomavirus 16; Cryo-electron Microscopy; Mass Spectrometry; Molecular Dynamics; Intrinsically Disordered Proteins; Virus Host Interactions
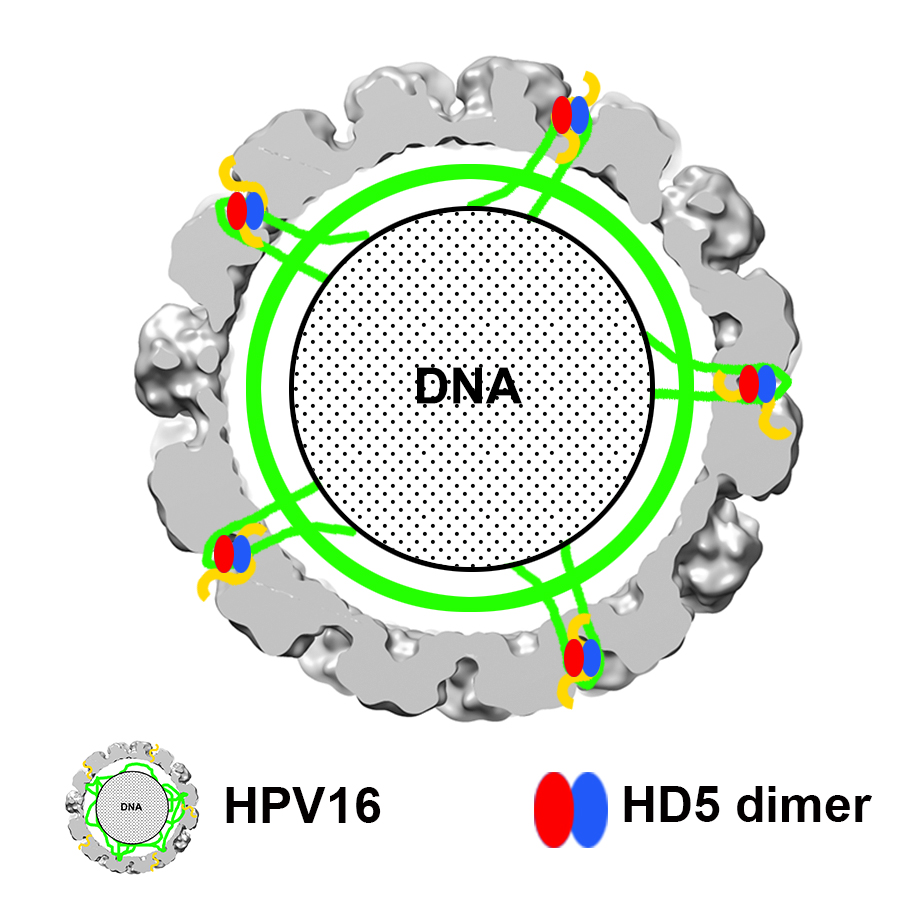
Graphical Abstract:
Human papillomavirus (HPV) infections are a major health burden. HPV is the most common sexually transmitted infection, causing a range of conditions including genital warts and cancer. While most HPV infections do not cause major health concerns, persistent genital HPV infection is linked to nearly all cases of cervical cancer, in addition to other types of cancers in men and women [1, 2]. Worldwide, cervical cancer causes approximately 270,000 deaths annually, with the highest mortality rates in low- and middle-income countries [3]. Vaccines against certain strains of HPV, including those that cause cervical cancer and genital warts, are clinically available [4]. They are believed to function primarily by eliciting neutralizing antibodies, and the humoral response to HPV infection has been an area of intense research [5, 6]. However, recognition of HPV by the innate immune response and the role of innate responses in stimulating a potent adaptive response to HPV is less well understood [7].
Defensin antimicrobial peptides are effector components of innate immunity. They are directly antimicrobial but also function indirectly through immunostimulatory activity [8-12]. Both of these activities likely contribute to their role in host defense against bacterial, viral, and fungal infections. Human defensins are classified into 2 groups, α and β, based on structure and sequence. The α-defensins are further subdivided into myeloid and enteric, based on the cell types in which they are primarily expressed. We have focused on the activity of human α-defensin 5 (HD5), because it is expressed in the male and female genitourinary tract [13-15] and has been shown to potently block infection of multiple serotypes of HPV, including high-risk HPV16 [16-20]. HPV16 and HPV18 together cause about 70% of all cervical cancers and are the 2 highest risk types of HPV [1].
A broad mechanism of α-defensin neutralization of non-enveloped viruses has emerged from studies of adenoviruses, polyomaviruses, and papillomaviruses [8, 19]. Although they do not block and have in some cases been shown to enhance initial binding of viruses to cells, α-defensins perturb uncoating and prevent the DNA genomes of these viruses from reaching the nucleus. For adenoviruses, HD5 bridges the interaction between 2 capsid proteins, thereby stabilizing the viral capsid and completely blocking uncoating within the endosome [21-25]. JC polyomaviruses are also neutralized by HD5-induced viral stability, which leads to altered intracellular trafficking [26]. The mechanism of HPV16 neutralization is more complex in that there are at least 2 effects of HD5 on entry. First, cleavage of the minor capsid protein L2 at the cell surface is blocked [18]. Second, although the virus is internalized and partially uncoats, the genome remains aberrantly associated with the major L1 capsid protein and fails to traffic to the nucleus [19]. The only exception to this paradigm of viral stabilization is that HD5 and the myeloid α-defensin HNP1 inhibit BK polyomavirus primarily by aggregating the virus and preventing binding to host cells [27]. Although all of these antiviral functions rely on defensin binding to the capsid, the molecular features recognized by α-defensins and their localization on the capsids of these disparate viruses are largely unknown.
To address this gap in knowledge and to clarify the structural mechanism of HD5 neutralization of HPV, we have used cryo-electron microscopy (cryoEM) and mass spectrometry to investigate the effect of HD5 binding on the HPV16 structure. The HPV capsid is composed of 72 L1 pentamers, arranged so that some pentamers have 5 neighbors (pentavalent capsomers) and others have 6 neighbors (hexavalent capsomers), and an undefined copy number of L2. The capsid is stabilized by disulfide bonds between cysteine residues of neighboring L1 capsomers [28 ], and a recent near-atomic resolution cryoEM structure of HPV has provided pseudoatomic coordinates for the L1 portion of the HPV16 capsid (PDB: 5KEP) [29]. The HPV L2 protein co-assembles with L1 and serves multiple functions including DNA packaging [30, 31], facilitating escape of the L2/genome complex from late endosomes and trafficking of the genome during entry [32-37], and chromatin binding linked to translocation of the L2/genome complex across the limiting cellular membrane during mitosis [38, 39]. L2 is predicted, for the most part, to be an intrinsically disordered protein [38], and it is also mostly an internal capsid protein [40]; however, there are regions of L2 that are known to emerge from the virion during the cell entry process and become accessible on the capsid surface [41-43]. Consistent with these observations, Guan et al tentatively assigned diffuse density on the exterior capsid surface primarily on the hexavalent L1 capsomers to L2 [29]. However, the precise locations of L2 within the capsid remain unclear.
In contrast to our studies of human adenovirus serotype 5 in complex with HD5 [23, 25], density attributable to HD5 on the HPV16 capsid is not readily apparent by cryoEM. Rather, the most notable structural change in HPV16, when in complex with HD5, is the increased ordering of density immediately below the capsid. Mapping by mass spectrometry provides evidence for HD5 interactions with disordered or flexible regions of L1 and L2. Molecular dynamics (MD) simulations indicate that favorable electrostatic interactions can be formed between the L1 C-terminal tail and an HD5 dimer. Thus, the combination of cryoEM, mass spectrometry, and MD simulations suggest a mechanism whereby HD5 binding entangles L1 and L2 by cementing interactions between disordered regions, leading to a more regular association between the genome and the capsid in the presence of HD5. This finding explains the failure of L1 and L2 to dissociate during cell entry in the presence of HD5 [19].
HPV16 pseudovirus (HPV PsV) containing the major capsid protein L1 and minor capsid protein L2 were prepared as previously described [44-47]. Briefly, an HPV16 PsV seed stock was made by co-transfecting 293TT cells with plasmids encoding codon-optimized HPV16 L1 and L2 (p16L1L2, gift of Martin Muller, GCRC) and an eGFP reporter (pfwB, gift from John Schiller, NCI). Cleared lysate from this transfection contained mature PsV and was then used to infect additional 293TT cells. The cells were lysed, and ammonium sulfate was added to a final concentration of 25mM to improve maturation of the HPV16 PsV. The lysate was treated with benzonase (SigmaAldrich) and Plasmid Safe RNase (Epicentre). NaCl was added to a final concentration of 850mM, and the HPV16 PsV was purified by ultracentrifugation through an OptiPrep gradient. OptiPrep was removed from the HPV16 PsV via agarose gel filtration, and buffer was exchanged into Dulbecco’s phosphate buffered saline (DPBS) with 0.8M NaCl.
In preparation for cryoEM studies, the purified PsV sample (~400 ng/µL) was diluted 75-fold in DPBS with 0.5M NaCl at pH 7.4. A 19.5 µL sample of diluted PsV was combined with 0.5 µL of concentrated HD5 (400µM in water) to produce a mixture with a final concentration of 10µM HD5, which is sufficient to neutralize HPV16 under these conditions. For these experiments, synthesized linear HD5 peptide (CPC Scientific, Sunnyvale, CA) was subjected to thiol-disulfide reshuffling and purified to homogeneity by reverse-phase high-pressure liquid chromatography to generate folded HD5 [20]. The PsV/HD5 mixture was incubated on ice for 45 minutes, and 3 µL HPV16 PsV alone or HPV16 PsV with HD5 was then applied to glow-discharged Quantifoil 2 × 2 400 mesh holey carbon grids. Grids were blotted until nearly dry and rapidly frozen in liquid ethane using a manual plunger.
CryoEM micrographs were collected on an FEI Titan Krios 300 kV transmission electron microscope with a Direct Electron DE20 direct detector for a total electron dose of 6000 electrons/nm2. Micrographs were collected at 29,000 × magnification with a defocus range of 0.8 µm to 3.5 µm. The HPV16 PsV alone dataset included 3304 micrographs and the HPV16 PsV with HD5 dataset included 3868 micrographs. Frame alignment and radiation dose damage compensation of the micrographs was performed with the DE_process_frames software (Direct Electron).
Individual particles were selected in a semi-automated fashion using E2 Boxer in the EMAN2 software package [48]. Estimations of the defocus values for the micrographs were made using GCTF [ 49].
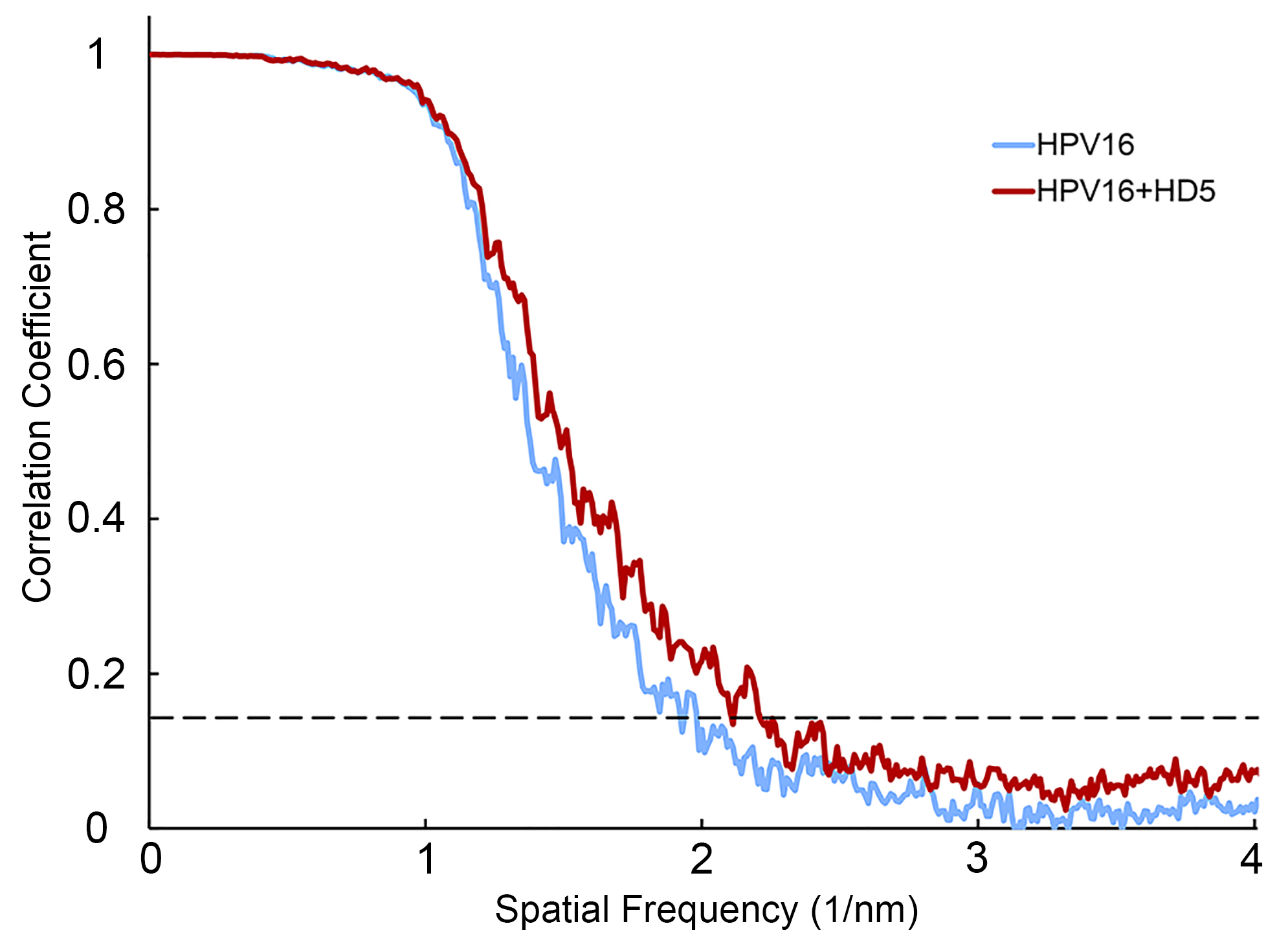
RELION 2.0 was used to extract individual particles from micrographs (0.126 nm/pixel or binned data at 0.252 nm/pixel) and for subsequent steps in refinement. A cryoEM structure of the HPV16 PsV (EMD: 5932) filtered to 6 nm resolution was used as the initial model for 3-D reconstruction [47]. This model was used to determine the orientation of each particle. A subset of particles was chosen through 2-D classification and 3-D classification for each structure. Binned data (0.252 nm/pixel) was used for 2-D classification and unbinned data (0.126 nm/pixel) was used for 3-D classification and refinement. Final refinement of the maps used 2900 particles for HPV16 PsV alone and 1742 particles for HPV16 PsV with HD5 using RELION 2.1. After refinement and postprocessing, the final resolutions of the HPV16 alone and HPV16+HD5 maps were 0.53 nm and 0.49 nm, respectively, as measured at the 0.143 threshold of a gold standard Fourier Shell Correlation plot. EMAN2 was used to low-pass filter the HPV maps to 2 nm resolution [48].
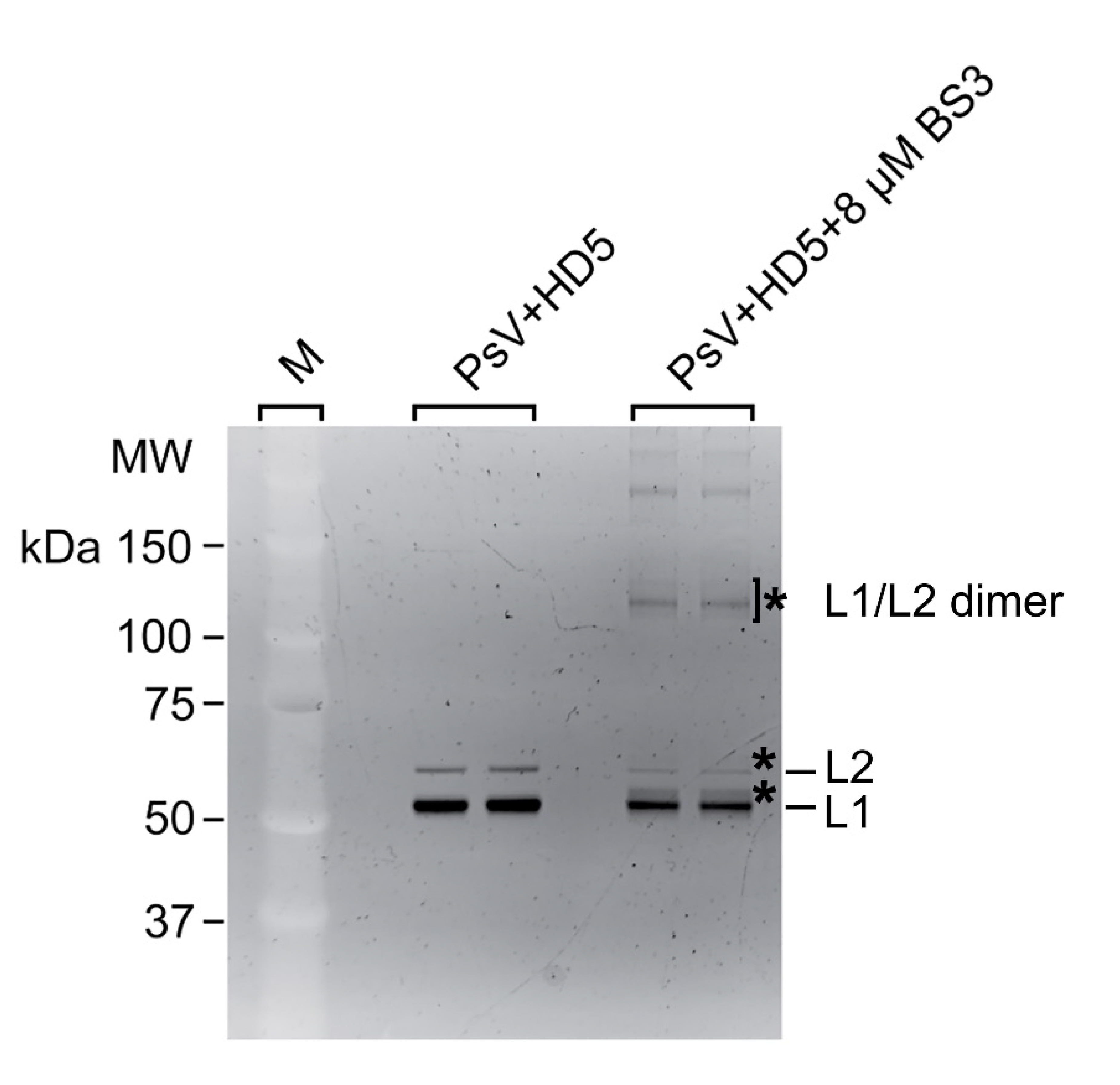
The complex of HPV16 PsV (1 µL containing 400 ng) and HD5-R13K/R32K [20] (2 µL containing 200 pmol) was mixed with 8μM bissulfosuccinimidyl suberate (BS3) and incubated at 25°C for 45 minutes in 10 µL of DPBS. The reaction was stopped by adding 1 µL of 130mM Tris buffer, pH 7.4. The reaction mixture was separated on a NuPAGE 4%–12% Bis–Tris gel using MES running buffer, and the crosslinked protein complexes were visualized by SYPRO Ruby. Three regions of the SDS-PAGE gel corresponding to crosslinked complexes for L1-HD5, L2-HD5, and the L1-L2 dimer (Figure S2) were excised, the proteins were in-gel digested using trypsin [50], and the digests were analyzed by LC-MS/MS as described below. Possible crosslinked peptides were identified using MassMatrix database search software (Version 3.10, MassMatrix, Columbus, OH), and all the possible crosslinked peptides were manually verified by inspecting their MS/MS spectra.
HPV16 PsV alone (1 µL containing 400 ng) and HPV16 PsV complexed with HD5 (2 µL containing 200 pmol) were treated with unlabeled- and 13C4-labeled-acetic anhydride (1 µL containing 100 nmol), respectively, in 10 µL of DPBS at 25°C for 1 hour. We also carried out a reverse experiment where the 13C4-labeled acetic anhydride was used to modify the HPV16 PsV alone sample and unlabeled acetic anhydride to modify the HPV16 PsV-HD5 complex sample. After the reaction, 2 μL of 1M hydroxylamine in water was added to the reaction mixture and incubated at 25°C for 30 minutes to stop the reaction as well as to reverse the acylation on tyrosine, cysteine, and histidine that might have occurred. Then, the samples treated with labeled and unlabeled acetic anhydride were mixed, transferred into an Amicon Ultra-0.5 10K centrifugal filter device, and the buffer salts, the reagents, and Optiprep in the sample were removed by repeated addition of 8M urea in 100mM ammonium bicarbonate and centrifugation at 14,000 g. The proteins were reduced by dithiothreitol (DTT) and S-alkylated by iodoacetamide; then the urea concentration was reduced to <2M and the proteins digested by trypsin in the filter device. The digest was analyzed by LC-MS/MS as described below. Lysine-acetylated peptides were identified using MassMatrix database search software. Carbamidomethylation of cysteine was set as a fixed modification, whereas variable modifications included 12C2-acetyl-lysine, 13C2-acetyl-lysine, and oxidation of methionine to methionine sulfoxide. The mass tolerance was set at 10 ppm for precursor ions and 0.8 Da for product ions. Strict trypsin specificity was applied. The ratio between a 12C2-acetyl-peptide and the same peptide carrying a 13C2-acetyl-group was manually calculated as previously described [51].
The digests prepared above were analyzed by data-dependent LC-MS/MS with collision-induced dissociation (CID) using a Thermo Fisher Scientific Fusion Lumos mass spectrometry system [52]. The HPLC column was a Dionex 15 cm × 75 μm id Acclaim Pepmap C18, 2 μm, 10 nm reversed-phase capillary chromatography column. The peptides were eluted from the column by an acetonitrile/0.1% formic acid gradient at a flow rate of 0.3 μL/min.
The mass spectrometry-derived peak ratios indicating the extent of chemical acetylation of lysines in the HPV16 capsid protein in the presence and absence of HD5 were analyzed with the Wilcoxon Signed Rank statistical test. This test is useful for analyzing matched paired data sets with non-parametric distributions. The one-tailed version of this test was used as the experimental ratios were compared to an expected value of 1, which would indicate no difference in lysine acetylation in the presence or absence of HD5. This test requires a minimum of 5 data points, therefore lysine acetylation data for 2 L1 residues, K59 and K479, was excluded from Table 2. The lysine acetylation data for one L2 residue, K318, was excluded from Table 3 as it was found to be not statistically significant.
Table 1. Mass spectrometry detected BS3 crosslinks between L1 and HD5-R13K/R32K
|
|
|
|
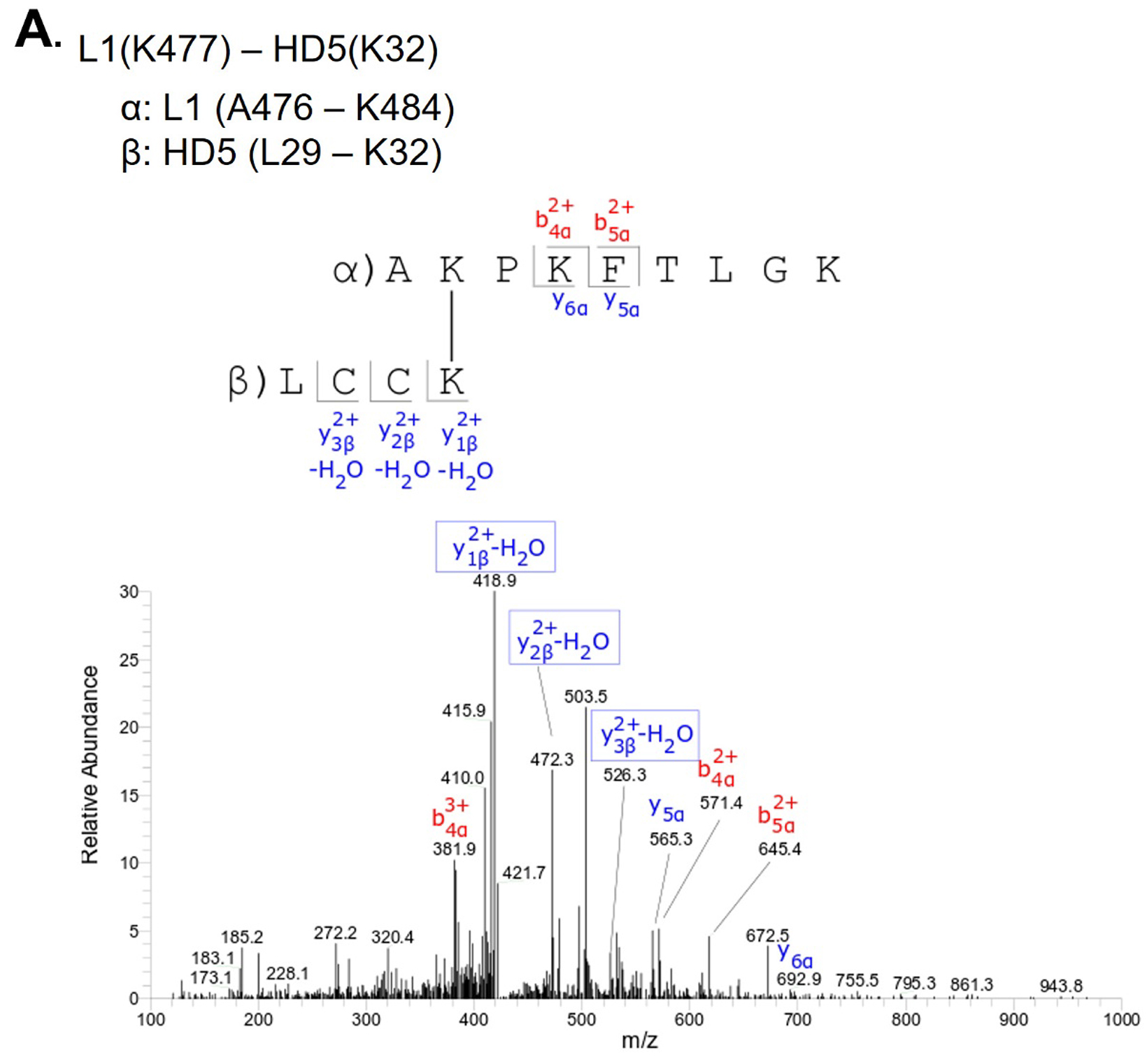
L1(K477) - HD5(K32) |
Above L1 band |
|
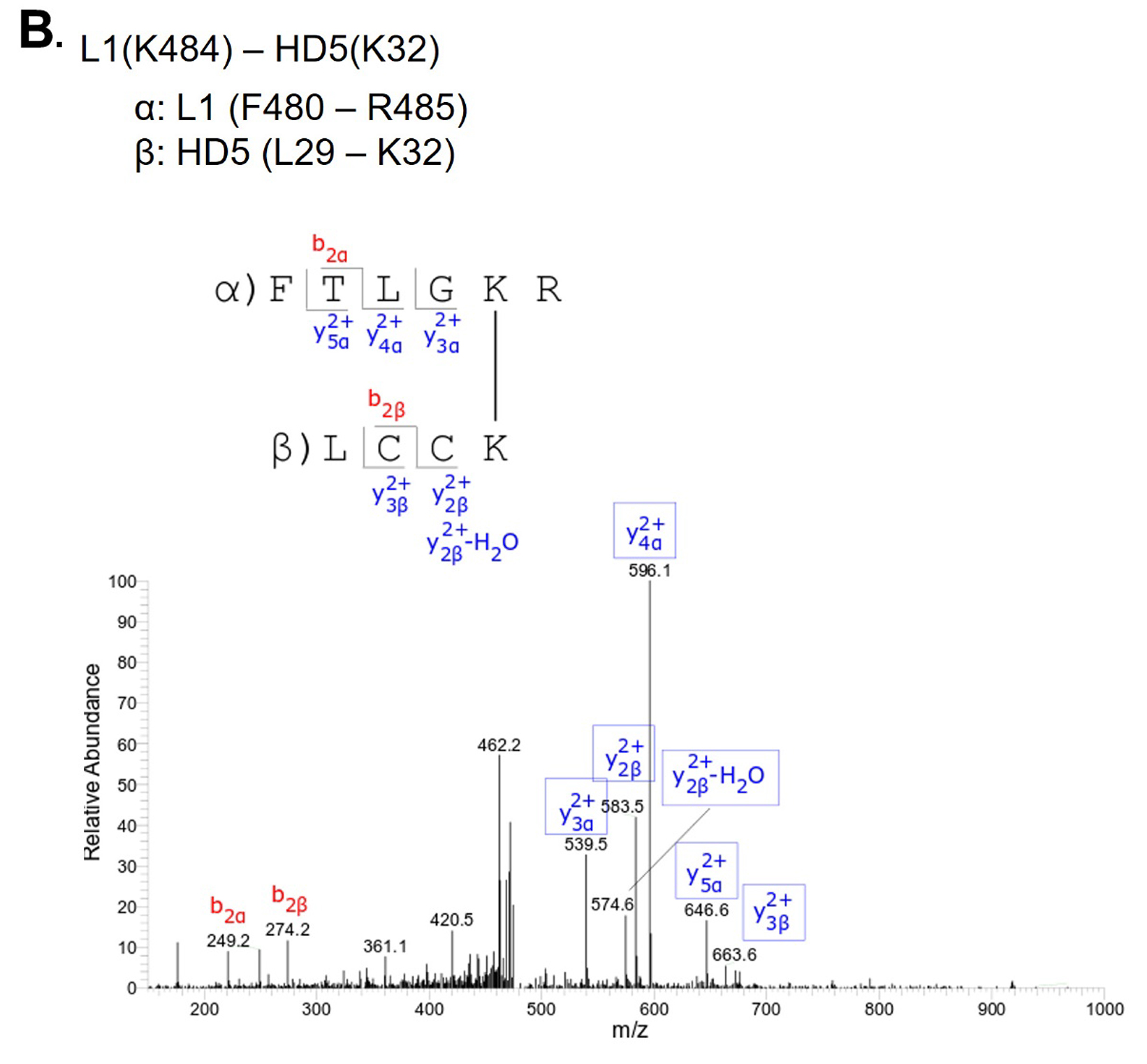
L1(K484) - HD5(K32) |
Above L1 band |
|
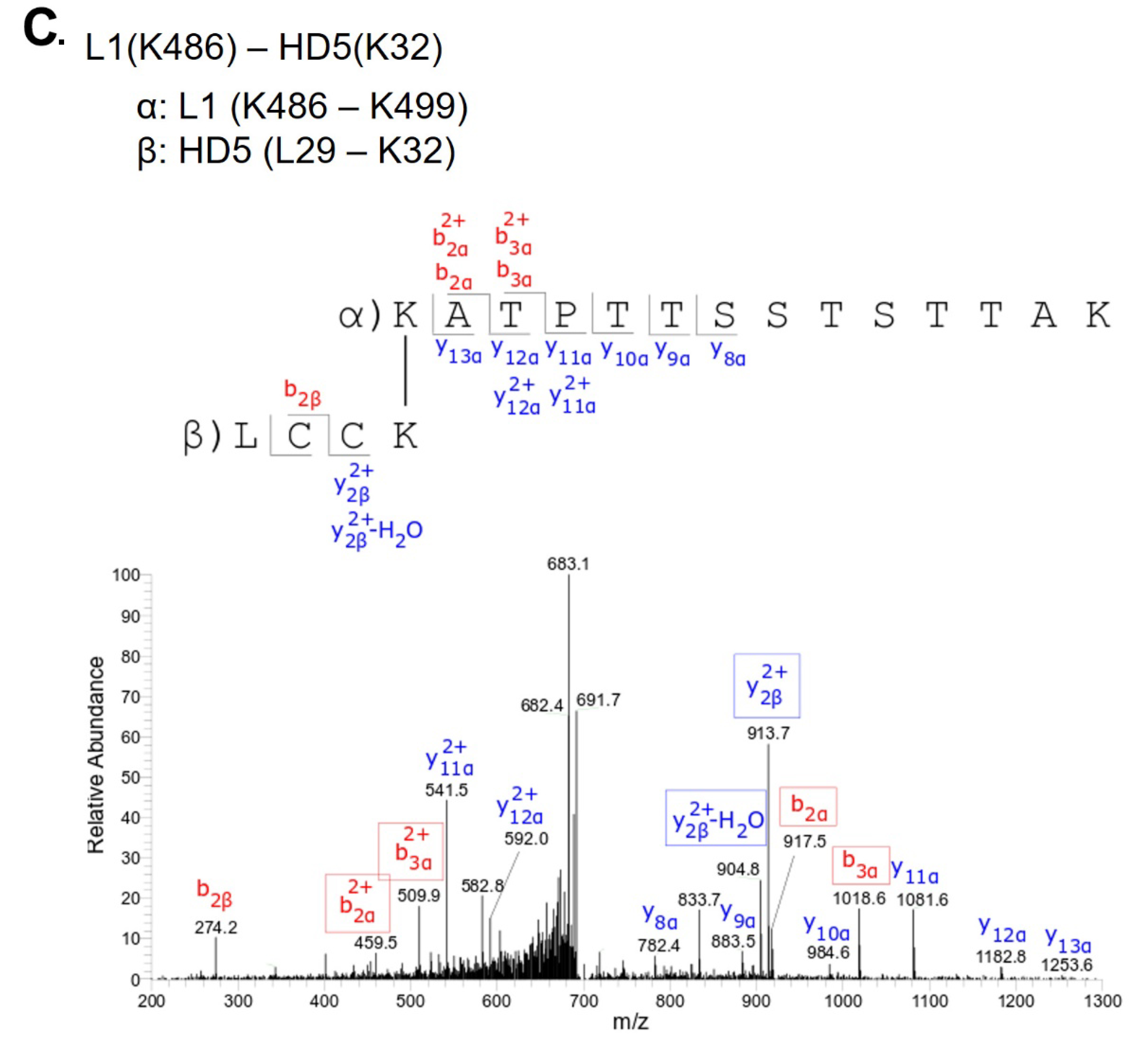
L1(K486) - HD5(K32) |
L1/L2 dimer region |
Table 2. Mass spectrometry detected lysine acetylation sites of L1 protein affected by the presence of HD5
|
Lysine Residue |
Mean Ratio (+HD5/-HD5) |
Standard Deviation |
Number of data points (n) |
P-value |
Effect of HD5 |
|
K430 |
0.36 |
0.05 |
5 |
0.05 |
HD5 reduces acetylation |
|
K437 |
0.78 |
0.10 |
6 |
0.025 |
HD5 reduces acetylation |
|
K442 |
0.66 |
0.17 |
6 |
0.025 |
HD5 reduces acetylation |
|
K467 |
1.25 |
0.12 |
6 |
0.025 |
HD5 enhances acetylation |
|
K475 |
0.72 |
0.06 |
6 |
0.025 |
HD5 reduces acetylation |
|
K484 |
0.62 |
0.18 |
6 |
0.025 |
HD5 reduces acetylation |
|
K486 |
0.71 |
0.24 |
6 |
0.025 |
HD5 reduces acetylation |
|
K499 |
0.73 |
0.24 |
6 |
0.025 |
HD5 reduces acetylation |
Table 3. Mass spectrometry detected lysine acetylation sites of L2 protein affected by the presence of HD5
|
Lysine Residue |
Mean Ratio (+HD5/-HD5) |
Standard Deviation |
Number of data points (n) |
P-value |
Effect of HD5 |
|
K20 |
0.25 |
0.26 |
5 |
0.05 |
HD5 reduces acetylation |
|
K309 |
0.75 |
0.24 |
6 |
0.025 |
HD5 reduces acetylation |
The UCSF Chimera [53] “Build Structure” and “Adjust Torsion” commands were used to build the starting conformation of an extended model for the C-terminal tail of 1 L1 subunit in PDB: 5KEP (chain F, modeled residues T481-L505). The backbone torsion angles of the modeled region were adjusted so that the C-terminal tail would pass through a capsid opening and extend away from the capsid. Minor backbone torsion angle adjustments were made with the Swiss-PDB Viewer Ramachandran Plot tool [54]. The C-terminal tail model was validated with the RAMPAGE Ramachandran plot analysis website [55] and found to have all residues in favored regions. The UCSF Chimera “Find Clashes/Contacts” command was used to check for clashes with the surrounding L1 chains.
To model the possible interactions between this extended model of the L1 C-terminal tail and an HD5 dimer, MD simulations were performed with NAMD [56] using implicit solvent and the CHARMM27 force field. Input coordinates were generated with 1 L1 Chain F (aa 8-505) and an HD5 dimer (PDB: 1ZMP) with 10 different starting positions for HD5 relative to the L1 C-terminal tail. MD simulations (5 nanoseconds) were performed, and nonbonded energies between the L1 Chain F and the HD5 dimer were calculated for the ending coordinates using the NAMD Energy plugin in VMD [57]. The MD simulation with the most favorable nonbonded interaction energy (-322 kcal/mol) at the end of the simulation was selected. This interaction energy is predominantly due to electrostatic interactions (-227 kcal/mol) with a smaller van der Waals component (-94 kcal/mol). The MD simulations were performed on the Case Western Reserve University High Performance Computing Cluster.
CryoEM maps for the HPV16+HD5 complex and HPV16 have been deposited in the EM database (www.emdatabank.org/) with respective accession numbers EMD: 20218 and 20219.
Samples of HPV16 pseudovirus (PsV) and HPV16 PsV in complex with a neutralizing concentration of HD5 (10µM) were vitrified, and cryoEM data collection was performed on a Titan Krios with a Direct Electron DE20 detector. Motion-corrected and dose-compensated micrographs of both samples showed well-dispersed particles with similar morphology: spherical capsids and prominent protrusions for the capsomers (Figure 1A). HPV16 PsVs have diameters of ~60 nm, consistent with previous structures [29, 47]. However, the viral capsids in the cryo-electron micrographs are not completely uniform, with heterogeneity in both size and shape (Figure 1A, arrows). Approximately 26% of particles in both the HPV16 and HPV16+HD5 datasets were observed to have either a non-round shape or a diameter smaller than 60 nm. The presence of HD5 did not affect the fraction of particles that were non-spherical. REgularized LIkelihood OptimizatioN (RELION) was used to sort the populations of HPV16 and HPV16+HD5 particles to include only those with spherical shape and a uniform diameter to produce a more homogenous dataset for reconstruction [59]. The reconstruction of HPV16 resulted in a 0.53 nm structure, while HPV16+HD5 resulted in a 0.49 nm structure (Figure 1B), as determined using gold standard Fourier shell correlation (FSC) curves at the 0.143 threshold (Figure S1). At this resolution, both HPV16 and HPV16+HD5 reveal α-helical features (Figure 1C) and have similar capsid structures. Subtraction of the HPV16 structure from the HPV16+HD5 structure did not result in difference density in the capsid that might be attributed to HD5 binding. Therefore, we hypothesized that HD5 binds to flexible or heterogeneous regions on the HPV16 capsid, which are not resolved.
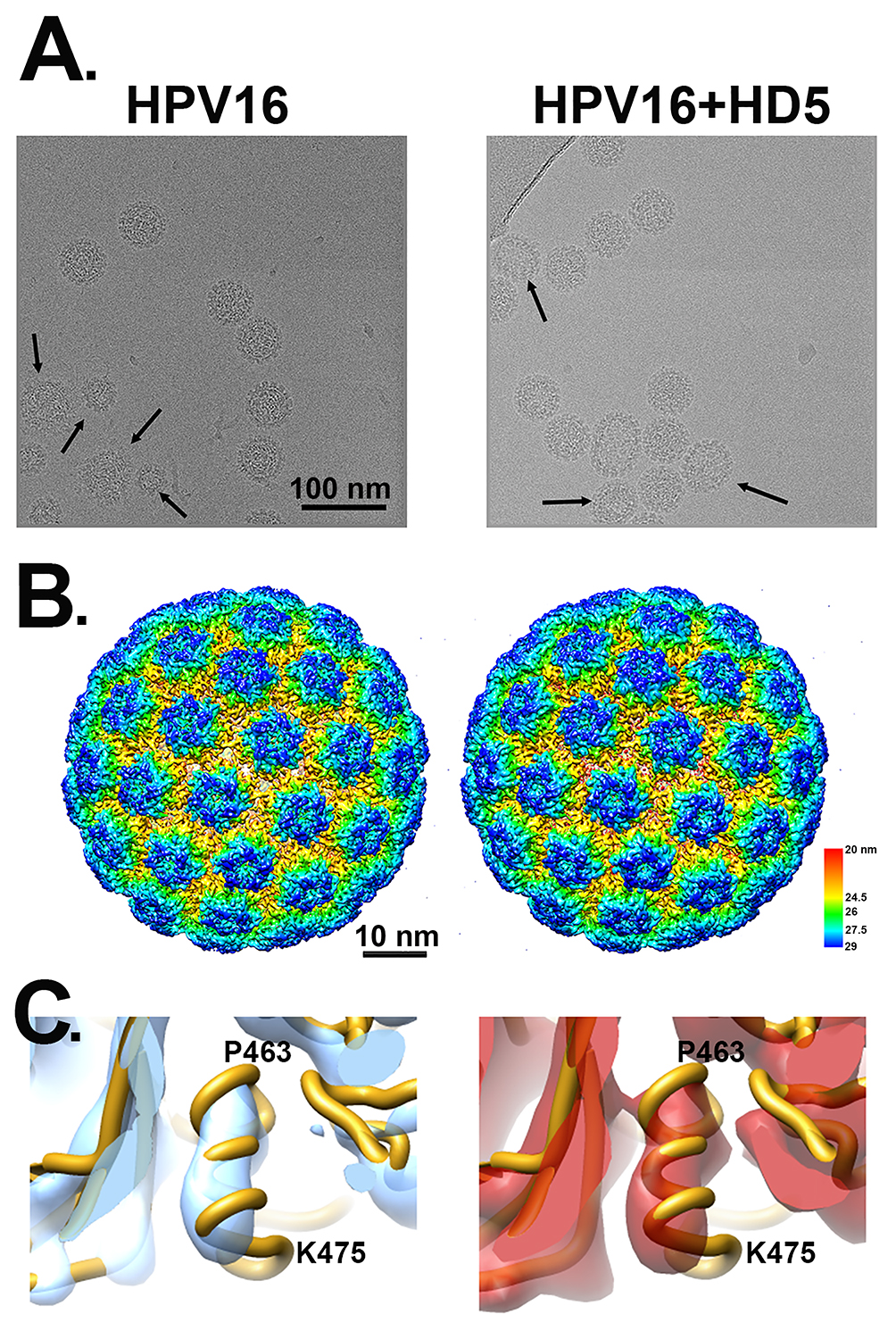
Figure 1. CryoEM of HPV16 and HPV16+HD5. (A) Representative cryo-electron micrographs of HPV16 (left) and HPV16+HD5 (right). Arrows point to capsids with heterogeneous size or shape. Scale bar = 100 nm. (B) Radially colored representations of HPV16 (left) and HPV16+HD5 (right). Scale bar = 10 nm. (C) Enlarged views of the HPV16 (left) and HPV16+HD5 (right) cryoEM maps showing α-helical density in L1 (aa 463-475, chain E) in both structures.
Prior cryoEM studies of HPV16 at pseudoatomic resolution indicate that the N- and C-terminal regions of L1 are disordered [29, 47]. In the high resolution cryoEM study by Guan et al, the 6 L1 chains in the asymmetric unit are slightly different and are missing between 2 and 17 residues at the N-terminus and between 19 and 25 residues at the C-terminus. No density is observed for the L1 termini in either the HPV16 or HPV16+HD5 cryoEM structures presented here, indicating that they remain disordered in the presence of HD5. In addition, like the near-atomic resolution cryoEM structure of HPV16 by Guan et al, the HPV16 cryoEM structures presented here do not reveal any well-defined density for L2. While surface-exposed regions of L2 and disordered N- and C-terminal tails of L1 could be involved in HD5 binding, these interactions might not result in any obvious density within the HPV16+HD5 cryoEM structure.
To investigate the possibility that HD5 binds to protein regions on the capsid that have a limited degree of flexibility, we filtered the cryoEM density map, resulting in lower resolution but a higher signal-to-noise ratio for regions of limited mobility. However, even filtering the HPV16 and HPV16+HD5 structures to 1, 1.5, and 2 nm still did not reveal any obvious density for HD5 on the capsid (Figure 2A). The cryoEM-derived L1 capsid coordinates fit well within both the HPV16 and HPV16+HD5 structures filtered to 2 nm resolution leaving no unexplained density that might be assigned to L2, the termini of L1, or HD5 (Figure 2B). However, we noted that the internal core density for HPV16+HD5 was different from that observed for HPV16 (Figure 2C). Thus, although there was no discernible change in capsid protein density, we did observe an effect of HD5 binding on the virion structure.
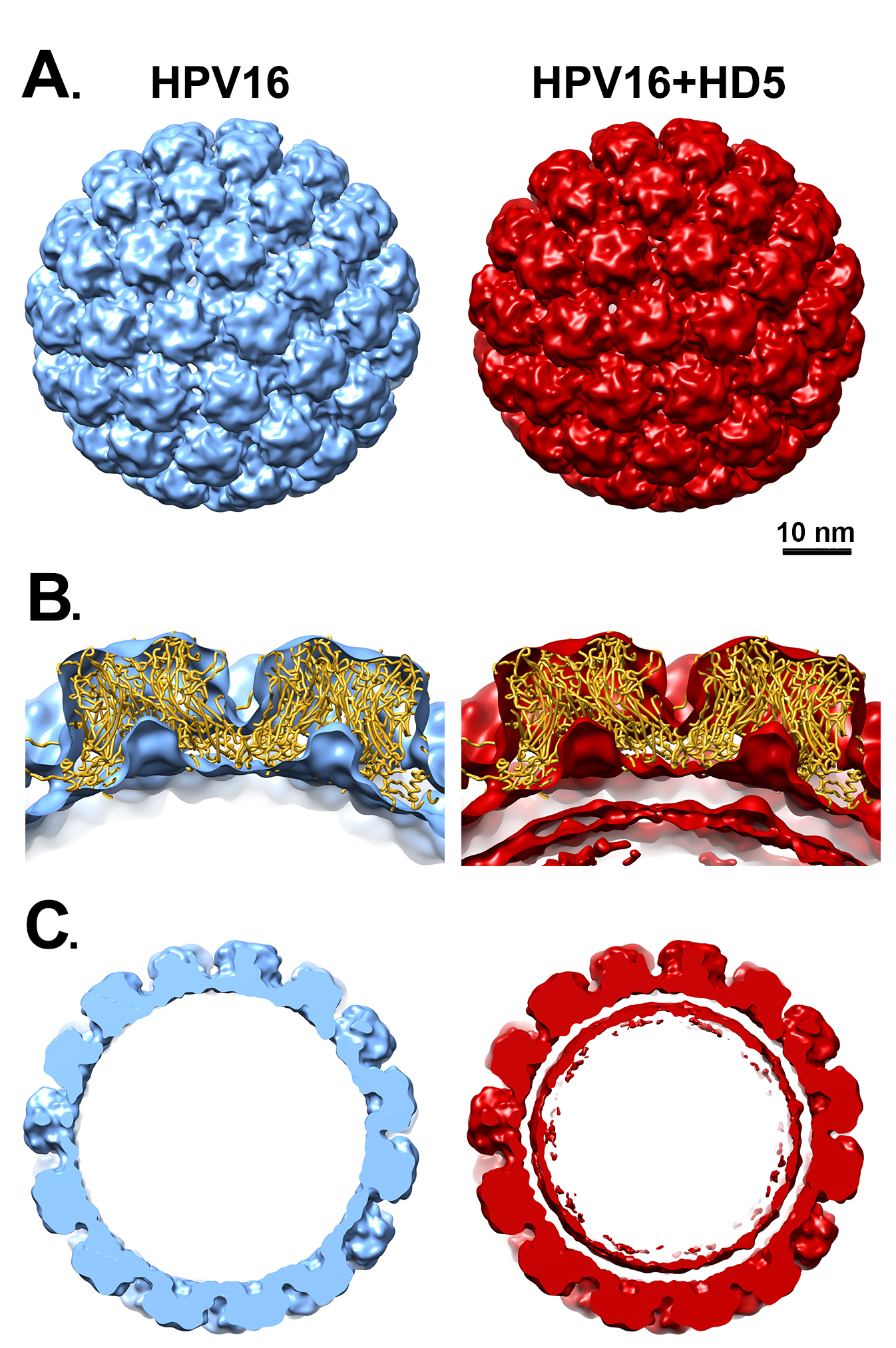
Figure 2. Filtered maps of HPV16 and HPV16+HD5. (A) Both cryoEM structures are shown filtered to 2 nm. Scale bar = 10 nm. (B) Cross-sectional views with docked L1 capsid coordinates (PDB: 5KEP) shown as a ribbon (gold) and oriented with the hexavalent L1 pentamer on the left and the pentavalent L1 pentamer on the right. In the HPV16+HD5 structure, additional density is observed below the L1 capsid. (C) Central slabs (10 nm thick) of both structures. Note the prominent internal core density below the L1 capsid in the HPV16+HD5 structure.
The HPV core includes portions of L2 and the genome, or in the case of PsV, randomly incorporated DNA. There is no evidence to suggest that the core of HPV16 has a regular structure, and application of icosahedral symmetry during the reconstruction process leads to most of the core density being averaged away. Accordingly, little to no core density is observed in the near-atomic resolution cryoEM structure of HPV16 by Guan et al [29] or in the near-atomic resolution HPV16 cryoEM structure presented here. Thus, the increased core density in the filtered HPV16+HD5 structure is striking and suggests a specific effect of HD5. A simple explanation might be that HD5 has penetrated the capsid and is bound to flexible regions of L1 or L2 or to the DNA inside the capsid. Although there are small openings in the HPV16 capsid (up to ~1.5 nm in diameter, Figure 3A), even an HD5 monomer (dimensions ~2 × 2.5 × 3.5 nm) is too large to enter into the core of the virion. The HD5 monomer contains 3 intramolecular disulfide bonds (Figure 4A). A linear form of HD5 without disulfide bonds might be able to enter the interior of the virion, but the properly folded, disulfide-bonded form of HD5, as used in our HPV16 + HD5 cryoEM study, would likely be restricted to binding on the capsid exterior. Also, antiviral activity of HD5 against HPV16 requires dimerization [20], and the HD5 dimer (~2.5 × 4 × 4 nm) is even more unlikely to pass through the small openings in the HPV16 capsid. Therefore, we conclude that HD5 does not enter into the interior of the virion, that the effect of HD5 on the HPV16 core is indirect, and that HD5 binding occurs on the exterior surface of the capsid.
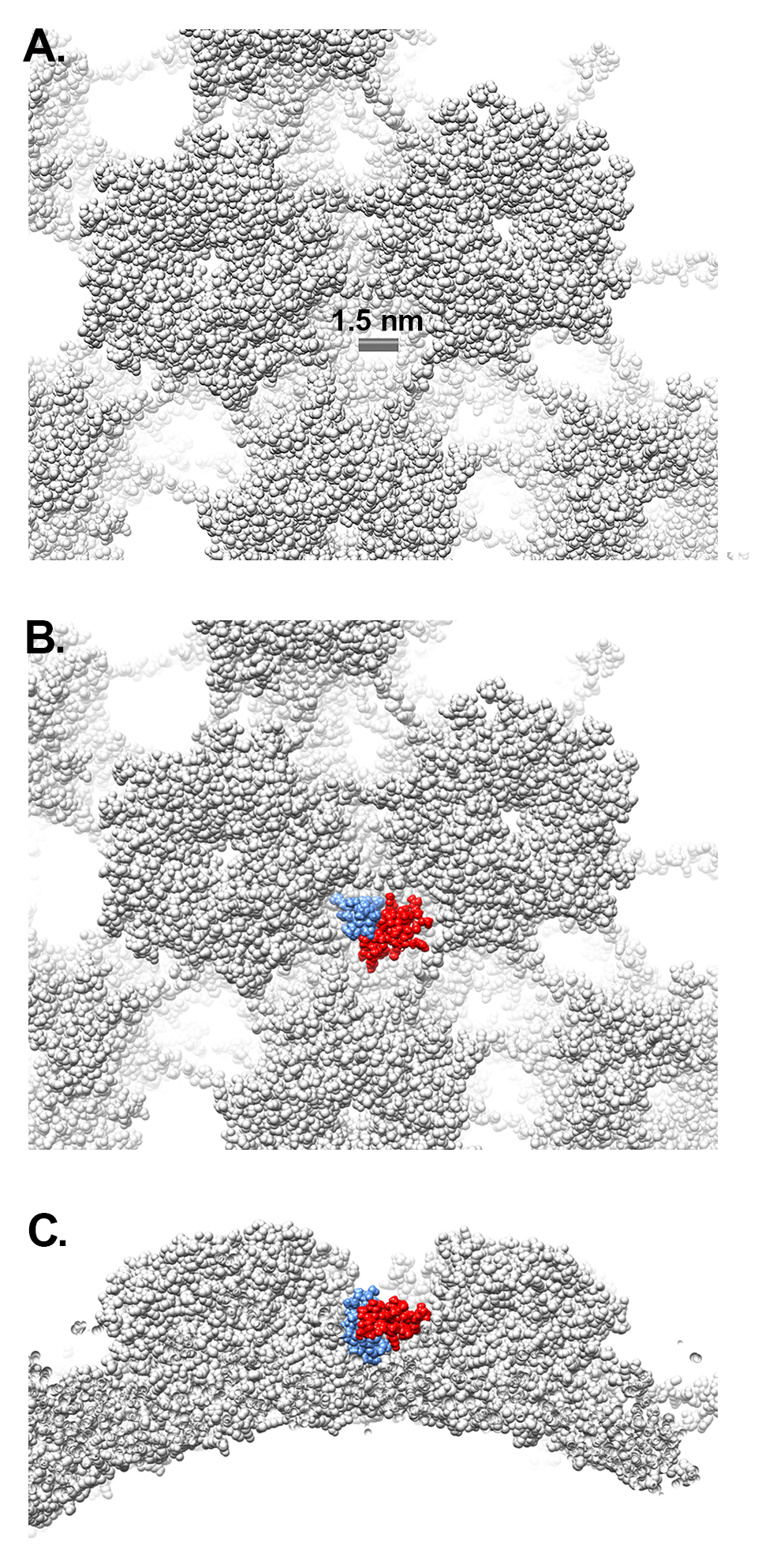
Figure 3. Gaps in the L1 capsid are smaller than HD5. (A) Space-filling representation of the L1 capsid with a scale bar indicating a capsid gap ~1.5 nm in diameter (PDB: 5KEP). (B) Space- filling representation of an HD5 dimer (red and blue) (PDB: 1ZMP, chains A and C) positioned within a depression between L1 pentamers. (C) Side view of panel B.
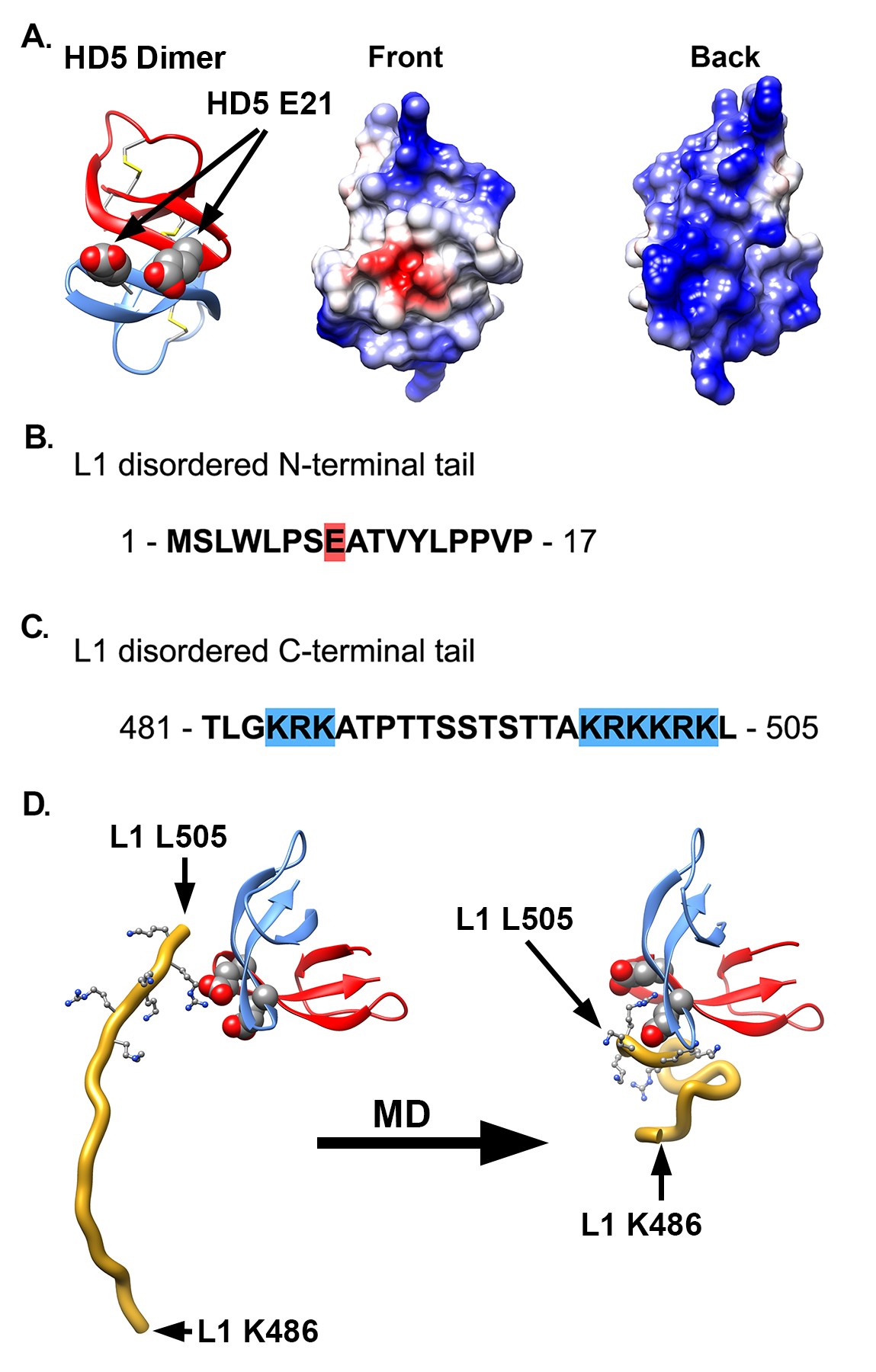
Figure 4. Proposed interaction between HD5 dimer and L1 C-terminal tail. (A) Ribbon and Coulombic surface representations of an HD5 dimer (PDB: 1ZMP, chains A and C). Disulfide bonds are depicted in stick representation and E21 residues in space-filling representation. The 2 E21 residues are next to each other in the dimer and form a negatively charged patch (red) on the Coulombic surface of a predominantly positively charged Coulombic surface. (B) Sequence of the longest disordered L1 N-terminal tail (aa 1-17) in the near-atomic resolution cryoEM-based HPV16 structure (PDB: 5KEP). (C) Sequence of the longest disordered L1 C-terminal tail (aa 481-505). In panels B and C charged residues are indicated with red (negative) or blue (positive) highlights. (D) MD-derived model of the interaction between an HD5 dimer and the L1 C-terminal tail (aa 486-505, gold ribbon). Both the starting and ending coordinates are shown. The negatively charged residues of the HD5 dimer (space-filling representation) form favorable electrostatic interactions with positively charged residues in the L1 C-terminal tail (499-KRKKRK-504, ball and stick representation).
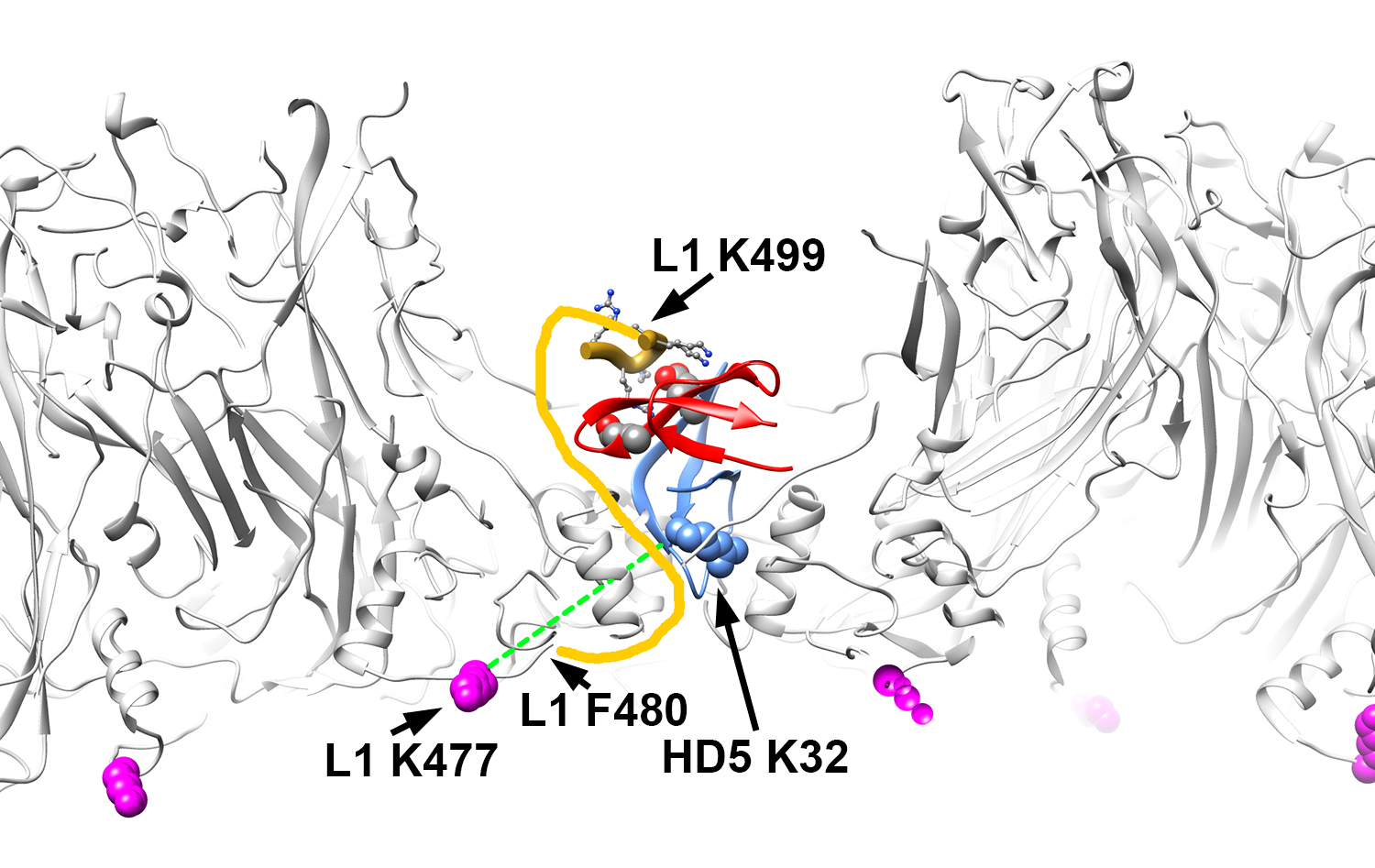
Figure 5. Structural model illustrating BS3 crosslinks between L1 and HD5 lysine residues. Side view of the L1 capsid (gray) with an HD5 dimer (red and blue) positioned within a depression between L1 pentamers such that one HD5 K32 residue (blue, space-filling) is directly above a capsid opening. The MD-derived model of an HD5 dimer and the last 6 residues of the L1 C-terminal tail (aa 499-505, gold ribbon) are shown as in Figure 4D. The flexible connecting residues between L1 F480 (Chain F) on the inner capsid surface and L1 K499 near the HD5 interaction site are depicted as a yellow line. The distance that would have to be spanned to connect these 2 residues (F480 and K499) is 3.7 nm, which is considerably less than the extended length of a 19 aa peptide (6.9 nm). Residue L1 K477 (magenta, space-filling) is detected as crosslinked to HD5 and is on the inner capsid surface. The dashed green line represents a distance of 2.8 nm and serves to indicate that the maximum distance constraint (3 nm) between Cα atoms of BS3 crosslinked lysines is met by this structural model.
During HPV entry, the virus uncoats, resulting in separation of most of L1 from a complex of L2 and genome [60-62]. Cell culture studies indicate that a major mechanism for HD5 inhibition of HPV16 infection is to prevent this separation [19]. If HD5 binds to disordered and highly flexible regions of both L1 and L2 on the exterior of the capsid, this would effectively entangle the 2 proteins and cement interactions between disordered regions of L1 and L2, leading to reduced degrees of freedom for L2 and a tighter association between L1 and the core components (L2 and DNA).
To investigate this mechanism, we performed crosslinking studies with BS3 to identify lysine residues in the vicinity of HD5 binding sites on the HPV16 capsid. For these studies, we used a mutant HD5 containing substitutions of 2 arginine residues (R13 and R32) for lysine (HD5-R13K/R32K), thereby introducing 2 additional primary amines for crosslinking. The HD5-R13K/R32K analog retains near wild-type antiviral activity against HPV16 PsV, unlike other HD5 analogs tested [17, 20]. Crystal structures of multiple HD5 analogs have been determined to have the same fold as wild-type HD5 [63-66], and the mass and HPLC retention time of the HD5-R13K/R32K analog during production and purification indicated that its fold is equivalent to wild-type HD5. HPV16 PsV was incubated with 20µM HD5-R13K/R32K to allow binding. The complex was incubated with 8µM BS3 followed by SDS-PAGE, resulting in the formation of multiple species with altered mobility compared to control (Figure S2). Three regions were excised from the SDS-PAGE gel, digested by trypsin, and analyzed by data-dependent LC-MS/MS with collision-induced fragmentation (CID) (Figures S3, S4, and S5). MS/MS analysis confirmed inter-protein crosslinks between L1 and HD5-R13K/R32K (Table 1), as well as intra-protein crosslinks within L1 (Table S1) and within HD5-R13K/R32K (Table S2). Because HD5-R13K/R32K has near wild-type antiviral activity, and the 2 substitutions of arginine to lysine are relatively conservative, the MS/MS finding of crosslinks between HD5-R13K/R32K and the L1 C-terminal tails should also be relevant for wild-type HD5. The observed crosslinks between L1 and HD5 all involve L1 residues near the C-terminus, K477, K484, and K486.
An additional mass spectrometry experiment was conducted to assess which regions of the HPV16 capsid are shielded by HD5 from chemical lysine acetylation. Unlabeled and 13C-labeled acetic anhydride samples were used to modify either HPV16 PsV alone or HPV16 PsV+HD5. Both forward (13C-labeled acetic anhydride with HPV16 PsV+HD5) and reverse (13C-labeled acetic anhydride with HPV16 PsV) experiments were conducted. The ratios of the 12C- and 13C-labeled mass spectrometry peaks were analyzed. A ratio equal to 1 would indicate no difference in lysine acetylation in the presence or absence of HD5. A ratio less than 1 would indicate that HD5 reduces acetylation at that site, while a ratio of greater than 1 would indicate that HD5 enhances acetylation at that site. Acetylation of 8 lysines in L1 and 2 lysines in L2 was found to be affected by the presence of HD5 (Tables 2 and 3). L1 lysine residues K430, K437, K442, K475, K484, K486, and K499, and L2 lysine residues K20 and K309, were all shielded by HD5 from chemical acetylation. For the remaining lysine, L1 K467, HD5 had the opposite effect and enhanced acetylation.
The simplest interpretation of the acetylation results for most of the L1 residues is that HD5 bound to the HPV16 capsid sterically blocks access of acetic anhydride to these sites, which is supported by the absence of HD5-induced conformational changes in the ordered region of L1. In contrast, the mass spectrometry results indicate that L1 K467 becomes more accessible to acetic anhydride after HD5 is bound to the virion, suggesting greater solvent accessibility for that particular residue in the presence of HD5. Since we observed no conformational changes in the ordered portion of the L1 capsid due to HD5 binding, the finding of enhanced acetylation at L1 K467 suggests a change in either L2 or the disordered termini of L1. Moderate resolution cryoEM reconstructions indicate that a portion of L2 is arranged within internal cavities of the L1 pentamers [40], and residue K467 is located in this vicinity within the internal cavity of the L1 pentamer. Thus, a shifting of L2 within the virion when HD5 binds could lead to greater exposure of L1 K467.
The differential lysine acetylation results also indicate that the presence of HD5 affects 2 lysine residues in L2 (Table 3). Both L2 K20 and L2 K309 show reduced acetylation in the presence of HD5. During cell entry, 12 residues at the N-terminus of HPV16 L2 are removed by cleavage by the host protease furin [67]. Thus, the N-terminal tail of L2 is at least transiently exposed on the virion surface [41,43, 68-72]. Protrusion of the N-terminal tail of L2 from a capsid opening near an HD5 binding site could explain why the presence of HD5 causes a comparatively strong (4-fold) reduction in acetylation of L2 K20 and would also be consistent with HD5-mediated inhibition of L2 cleavage by furin [18]. Multiple regions of L2, in addition to the N-terminal tail, emerge from the virion and become accessible on the capsid surface [42, 73]. Exposure of a middle segment of L2 through a capsid opening near an HD5 binding site could explain why the presence of HD5 causes a reduction in acetylation of L2 K309.
Based on the mass spectrometry results, we considered possible interactions between L1 and HD5. Examination of the crystal structure of an HD5 dimer (PDB: 1ZMP) indicates that the surface is mostly positively charged, with just 2 negatively charged residues (E21 from each monomer) forming a small negatively charged patch (Figure 4A) [74]. We previously identified E21 as one of the critical residues in HD5 for antiviral activity against HPV16 [17 ]. In addition, 2 additional critical residues, Y27 and L29, are at the HD5 dimer interface and may be important for maintaining the dimeric form of HD5. On the virus, the disordered N-terminal tail of L1 has only a single negatively charged residue (Figure 4B), while the disordered C-terminal tail has 2 regions of multiple positively charged residues (Figure 4C). The first ordered residues at the N-terminal end and the last ordered residues at the C-terminal end are positioned on the inner surface of the L1 capsid [29]. Potentially, both the disordered N- and C-terminal tails could protrude through gaps in the L1 capsid (Figure 3A). However, given that the C-terminal tail is both more hydrophilic and longer than the N-terminal tail, it seems more likely that the C-terminal tail is exposed on the exterior of the capsid. While exposure of the C-terminal tail may only be transient, it would still offer a potential binding site for HD5. Accordingly, we hypothesize that the most distal, positively charged region of the L1 C-terminal tail (499-KRKKRK-504) interacts with the negatively charged patch of an HD5 dimer formed by 2 copies of E21. The MD simulations with L1 and an HD5 dimer indicate that favorable electrostatic interactions can be formed (Figure 4D). Furthermore, modeling studies indicate that the disordered L1 C-terminal tail is long enough to protrude through openings in the HPV16 capsid. Interaction of HD5 dimers with L1 C-terminal tails protruding from the capsid would effectively tether HD5 within depressions between L1 pentamers of the HPV capsid (Figure 3B and C). Given the stretch of 6 positively charged residues near the L1 C-terminus, there could be multiple, electrostatically favorable, interactions formed between an HD5 dimer and L1. Both the disordered nature of the L1 C-terminal tails and the possibility of multiple distinct binding interactions between HD5 and L1 would make it likely that no cryoEM density would be observed for the L1 C-terminal tails and bound HD5 dimers within the capsid depressions in the HPV16+HD5 cryoEM structure. Therefore, although unproven, our models supported by MD simulations provide a plausible explanation for both the cryoEM and mass spectrometry results and a putative position for HD5 on the viral capsid.
We then determined whether positioning HD5 within depressions between L1 pentamers is consistent with our crosslinking mass spectrometry (CX-MS) results. We assumed a maximum distance of 3 nm between Cα atoms of BS3 crosslinked lysine residues [58]. We also noted that crosslinks only form on residues that are solvent accessible [75]. The CX-MS results indicate that a lysine on the inner capsid surface, L1 K477, is crosslinked to HD5. Since even an HD5 monomer is too large to pass freely through openings in the L1 capsid (Figure 3), we hypothesize that the chemical crosslinker must span the L1 capsid through a gap in the capsid. The cryoEM density of HPV16+HD5 indicates that the α-helix near K475 is still intact in the presence of HD5 (Figure 1C), suggesting no conformational change for this region of L1 in the presence of HD5. Positioning an HD5 dimer with one K32 residue directly above an opening in the L1 capsid places its Cα atom within ~2.8 nm of the Cα atom of the nearest L1 K477 on the inner capsid surface (Figure 5). The rest of the disordered L1 tail (yellow line in Figure 5) is sufficiently long (19 aa with a maximum extended length of 6.9 nm) to span the distance (3.7 nm) between the last ordered residue in the structure (F480) and the first residue supported by MD simulations to be in contact with HD5 (K499 in Figure 4D). Thus, this modeled HD5 position is feasible.
CX-MS indicates additional crosslinks between L1 (residues K484 and K486) and HD5 (Table 1). Since these 2 L1 residues (K484 and K486) are within the flexible connecting peptide between L1 F480 on the inner capsid surface and L1 K499 near the HD5 interaction site, it is difficult to measure the distance that would be spanned by a crosslinking molecule. For K484 and K486 to be within ~3 nm of HD5, the flexible L1 C-terminal tail likely bends up towards the surface of the virion, as we have modeled in Figure 5. Thus, with an HD5 dimer positioned within a depression between L1 pentamers and the L1 C-terminal tail protruding through a gap in the capsid, all of the cross-linked residues meet the spatial constraints of the experimental results.
The mass spectrometry acetylation data is also consistent with the structural model derived from the CX-MS and cryoEM results and MD simulations with an HD5 dimer positioned within a depression between L1 pentamers. Figure 6A, B, and C illustrate how an HD5 dimer bound to a protruding L1 C-terminal tail would restrict access to L1 residues K430, K437, K442, K475, and K484 in the HPV16 capsid. Two additional L1 residues with reduced acetylation in the presence of HD5 (K486 and K499) are within the flexible L1 C-terminal tail.
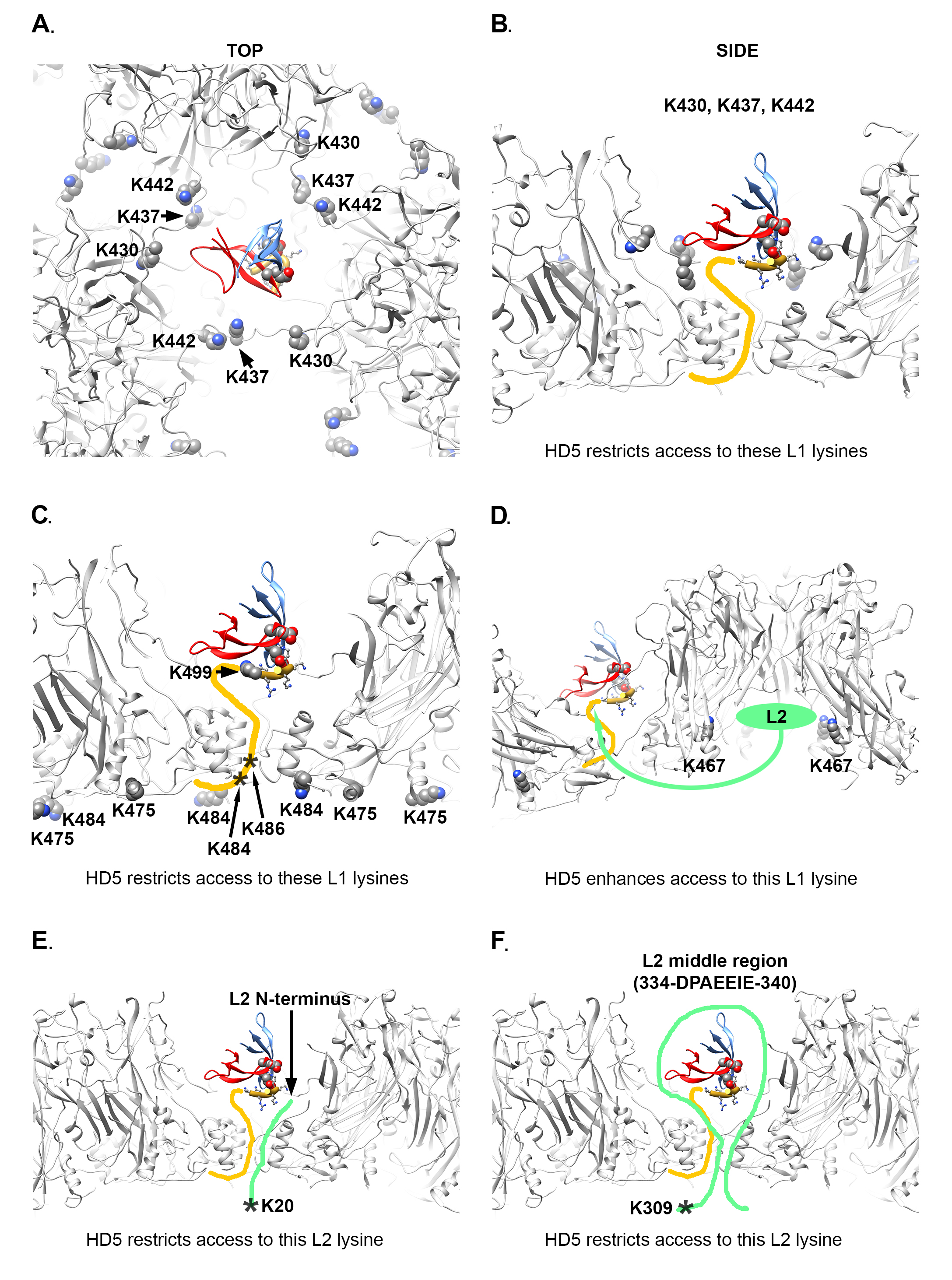
There is 1 L1 residue (K467) which displays enhanced acetylation in the presence of HD5. As mentioned above, residue K467 is located within the internal cavity of an L1 pentamer near an observed L2 binding site [40]. We propose that binding of HD5 to the HPV16 capsid induces a rearrangement of L2 within the virion which leads to increased solvent accessibility for L1 K467 (Figure 6D). We note that there is a negatively charged region within the HPV16 L2 sequence (Figure S6). Given that the HD5 dimer is mostly positively charged (Figure 4A), it seems plausible that the negatively charged region of L2 might shift its location within the virion after HD5 is bound.
Although there is no high-resolution structural information for L2, it is known that the L2 N-terminal region and various additional segments in the middle of L2 are transiently exposed on the surface of the virion [41-43, 68-73]. Even if L2 K20 remains inside the capsid, the presence of HD5 above the closest capsid opening could reduce access of acetic anhydride to this site (Figure 6E). In the case of L2 K309, this lysine residue is close to the negatively charged region (334-DPAEEIE-340) within the HPV16 L2 sequence (Figure S6). If the negatively charged region of L2 shifts its location within the capsid after HD5 is bound, this could plausibly block access of acetic anhydride to L2 K309 (Figure 6F).
The MD-derived model of the HD5 dimer and the C-terminal 6 residues of L1 is shown in the same orientation with respect to the HPV capsid in all panels of Figure 6, however this is a different orientation than shown in Figure 5. In Figure 6 the distance that would have to be spanned to connect L1 F480 on the inner capsid and L1 K499 in the MD-derived model is 3.2 nm, which is less than that in Figure 5, illustrating BS3 crosslinks between L1 and HD5 lysine residues. It is highly plausible that BS3 crosslinking would affect the position of the flexible L1 C-terminal tail and the bound HD5 dimer.
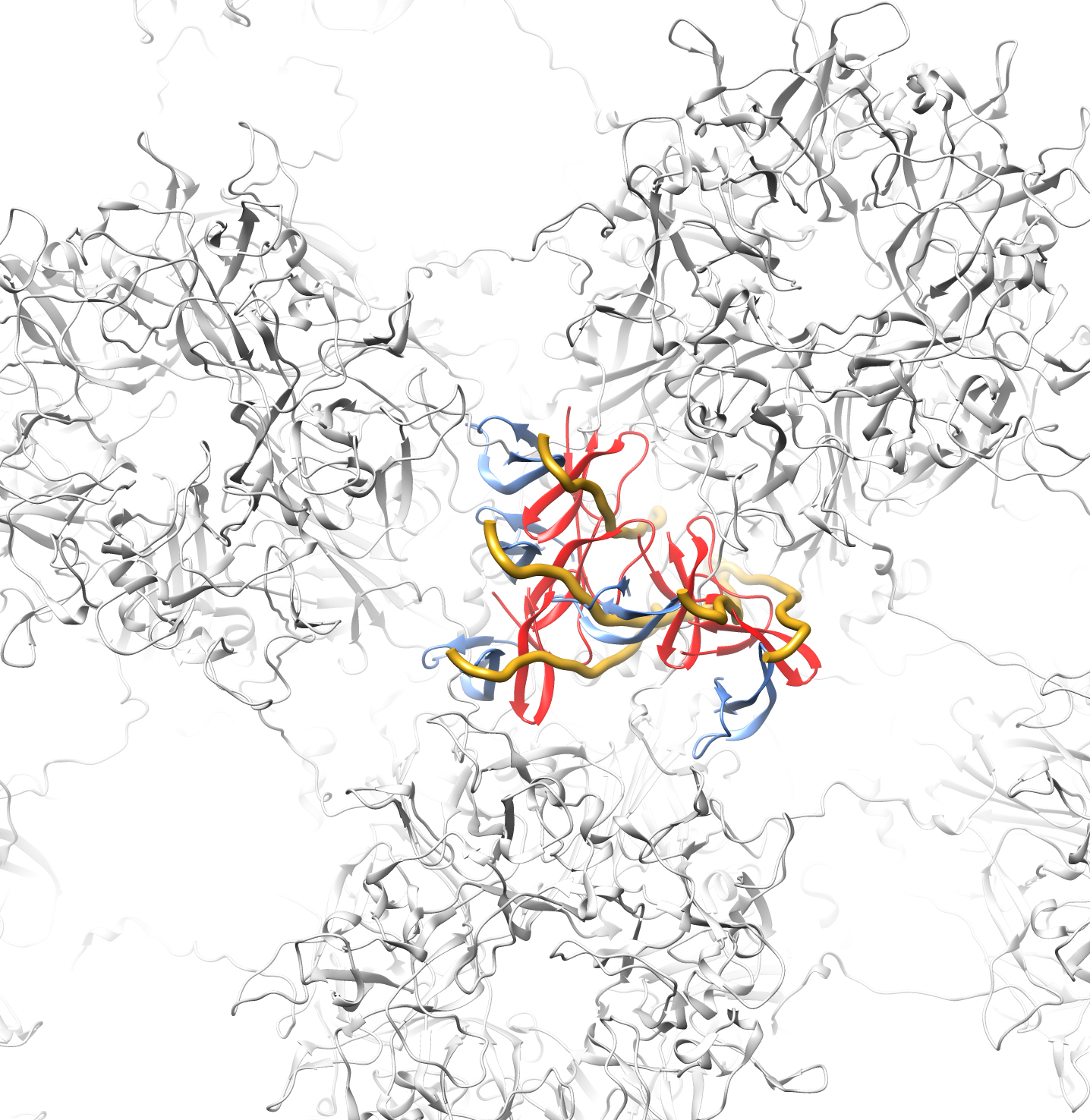
An overview of the differential lysine modification data for an L1 pentamer is shown in Figure 7, with the most highly affected lysine residue, K430, in magenta and the other affected lysine residues in green. It is notable that all of the affected lysines in the ordered portion of L1 are on the lower half of an L1 pentamer (Figure 7B). The proposed model for the HD5/HPV16 interaction places the most highly affected L1 residue (K430) in close proximity to HD5 (Figure 7A and B). The cryoEM structures of HPV16 and HPV16+HD5, the CX-MS data, and mass spectrometry acetylation data presented here are all in agreement with the proposed structural model with HD5 binding within a capsid depression and tethered to this site by a protruding disordered peptide of L1 and/or L2. It is important to note that tethering of an HD5 dimer to the HPV16 capsid via a disordered peptide would result in various possible positions for HD5 within the capsid depression (Figure 8). This heterogeneity in the HD5 position would explain why no cryoEM density was observed for HD5 in the HPV16+HD5 structure, even after filtering the HPV16+HD5 structure to 2 nm resolution (Figure 2).
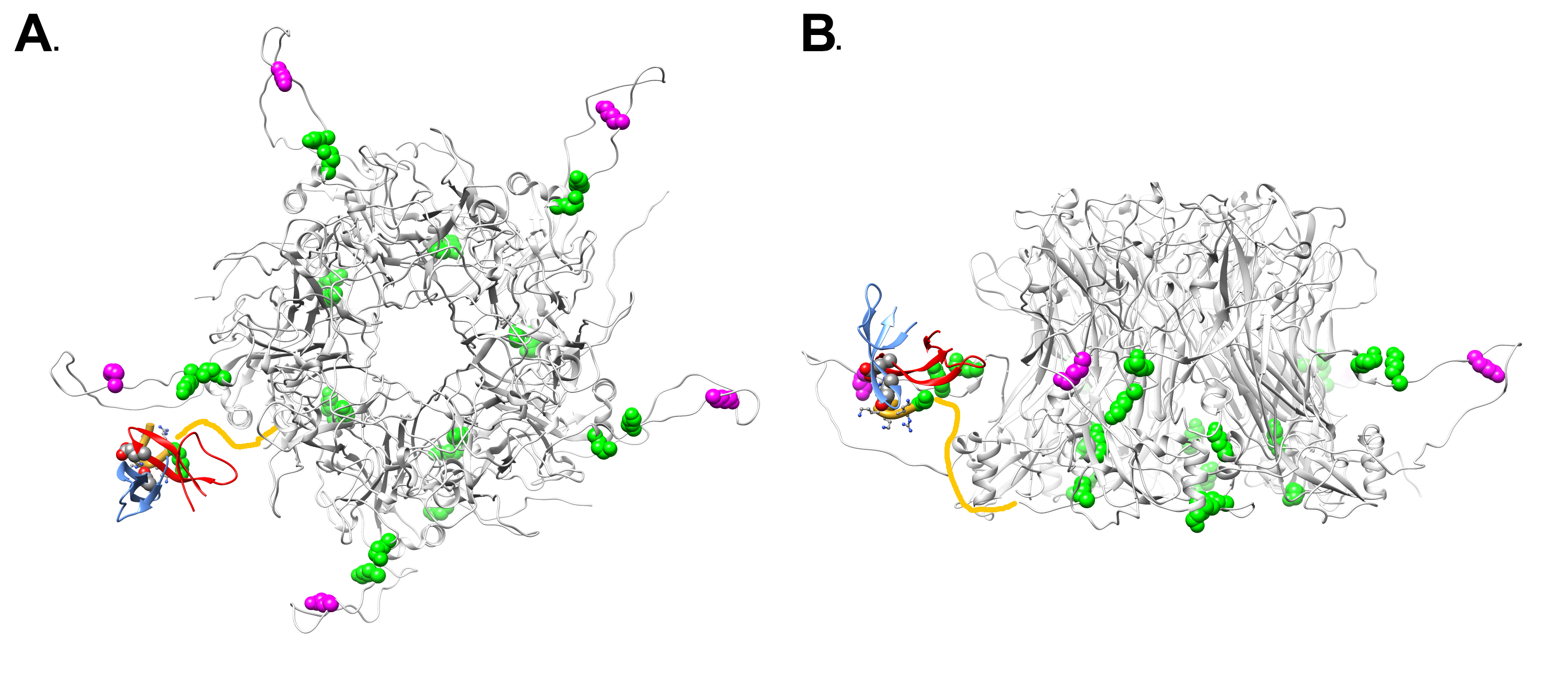
Figure 7. Overview of L1 lysine residues affected by HD5 with modeled position of HD5 dimer and L1 C-terminal tail. (A) Top view of an L1 pentamer (PDB: 5KEP, Chains B-F) with the lysine acetylation sites affected by the presence of HD5 in space-filling representation. The most affected lysine (K430) is in magenta and the other affected lysines (K437, K442, K467, K475, K484, K499) are in green. Some of the affected lysines are in the portion of the L1 C-terminal tail depicted as a yellow line (K484, K486). The MD-derived model of the HD5 dimer and the last 6 residues of the L1 C-terminal tail (aa 499-505) are in the same orientation with respect to the HPV capsid as in Figure 6. (B) Side view of panel A.
Combining experimental information from cryoEM and mass spectrometry has enabled us to propose a model for how HD5 affects HPV16 structure and cell entry. The cryoEM results indicate that HD5 does not bind homogeneously to the well-ordered region of the L1 capsid. Nevertheless, the cryoEM results do show the effect of HD5 binding in stabilizing the capsid/core interaction with increased core density in the HPV16+HD5 cryoEM structure (Figure 2). The pseudoatomic structure of the L1 capsid [29] reveals openings in the capsid of ~1.5 nm in diameter. While these openings are too small to enable HD5 to enter the interior of the virion, they are large enough to allow disordered N- or C-terminal tails of L1 and portions of the disordered L2 protein to protrude through the capsid and interact with HD5. Thus, we propose that HD5 dimers are tethered within capsid depressions by disordered C-terminal tails of L1 and unidentified flexible or disordered regions of L2. The variable nature of HD5 tethering to the HPV16 capsid via disordered peptides would explain why no density is observed for HD5 in the subnanometer resolution cryoEM structure of HPV16+HD5 presented here (Figure 1). The proposed interconnection of L1 and L2 via HD5 may lead to increased ordering of L2 or the genome below the L1 capsid and would explain why more density is observed below the capsid in the cryoEM structure of HPV16+HD5 (Figure 2). In the absence of HD5, the L2/genome complex may be only loosely associated with the L1 capsid leading to separation of the L1 capsid and the L2/genome complex by the appropriate cellular triggers during cell entry [76] (Figure 9A). In the presence of HD5, regions of L2 may be so tightly associated with HD5 and the L1 capsid that separation of the L1 capsid and the L2/genome complex is impeded (Figure 9B). In our model, with HD5 present, the L1 capsid only partially uncoats during cell entry and the virion is mostly held together by L1/L2/HD5 interactions. This partial uncoating in the presence of HD5 would allow antibodies to access the genome (anti-BrdU) and internal epitopes of L1 exposed by cathepsin-mediated degradation (33L1-7 antibody) [16, 19]. Importantly, the hyper-stabilized interaction of L1 with the L2/genome complex leads to viral neutralization [16, 19].
Our model is consistent with both the new mass spectrometry data presented here as well as our prior biochemical and infectivity studies. For example, it offers a possible explanation for the ability of HD5 to prevent furin cleavage of L2 [18]. Localization of HD5 within capsid depressions may provide steric hindrance for the interaction of furin with the N-terminal tail of L2. Either HD5 prevents furin from approaching close enough to the capsid surface to effectively cleave the L2 N-terminal tail, or more likely, HD5 blocks N-terminal tails of L2 from protruding far enough to be cleaved by furin. As furin is significantly larger than an HD5 dimer, it seems likely that the L2 N-terminal tail would have to protrude above the capsid depressions in order to be cleaved by furin. The finding that furin-cleaved HPV16 is still sensitive to neutralization by HD5 [19] can also be explained by our HD5/HPV16 interaction model. Furin-cleaved HPV16 would be able to form the same interactions with HD5 as we have proposed for uncleaved HPV16. Therefore, the same hyper-stabilization of L1 with the L2/genome complex would be predicted to form with furin-cleaved HPV16, leading to viral neutralization.
Confirming which molecular components account for the density observed below the capsid in the cryoEM structure of HPV16+HD5 is challenging for multiple reasons. First, this density is not resolved at high resolution and corresponds to heterogeneously organized components. Second, cryoEM studies of L1-only virus-like particles (VLPs) would not be expected to inform our model, since it has been shown that L2 is required for DNA packaging in HPV16 [30, 31]. It is anticipated that structures of L1-only VLPs, either in the presence or absence of HD5, would not show any core density. In addition, the absence of L2 in L1-only VLPs may expose non-physiologic HD5 binding sites that are not present in mature virus or in PsV composed of L1 and L2. Thus, any additional density found in L1-only VLPs in the presence of HD5 may not be relevant to PsVs used in infection studies [16-20]. Previous studies have shown that HD5 protects both L1 and L2 from trypsin cleavage [19]. Although this prior study does not provide any direct evidence regarding where HD5 is binding to the capsid, it does suggest that both L1 and L2 are involved in the interaction with HD5. In previous cryoEM analyses of adenovirus neutralization by HD5, we proposed that HD5 interacts with a short region at the base of the fiber and a portion of the large, RGD-containing loop of penton base [23, 25]. In both the adenovirus and HPV16 cases, the structural models involve interactions of HD5 with disordered peptide regions, which makes HD5/virion interactions challenging to study by structural methods. In this report we show that mass spectrometry can add information that is complementary to cryoEM, and that the combination facilitates the generation of structural models. However, as indicated in Figure 8, several models and orientations of HD5 relative to L1 and L2 are consistent with the experimental data. Although more precision may be precluded by the dynamic nature of the capsid and its interactions with HD5, this model will guide future work to confirm which regions of HPV16 L1 and L2 are directly involved in binding HD5. These structural analyses will lead to a greater understanding of the mechanism of HD5 neutralization and the role of defensins in immunity to HPV and other viral infections.
The authors gratefully acknowledge John Spear and Duncan Sousa at the Southeastern Consortium for Microscopy of MacroMolecular Machines for their assistance with acquisition of cryoEM data on the Titan Krios. We thank Sudheer Molugu at the Cleveland Center for Membrane and Structural Biology for his assistance in collection of preliminary cryoEM data and the Case Western Reserve University High Performance Computing staff for their assistance with campus computing resources. We thank Youngmee Sul and Angela Katsuyama at the University of Washington for confirming HPV16 PsV inhibition by HD5 under cryoEM conditions and for pilot studies of BS3 crosslinking. We also thank Belinda Willard and Ling Li of the Proteomics and Metabolomics Core at the Cleveland Clinic Lerner Research Institute for acquisition of the mass spectrometry data. At Case Western Reserve University we thank Sahil Gulati for assistance with the SYPRO Ruby stained SDS-PAGE gel, and Ming Li and Jean-Eudes Dazard for assistance with the Wilcoxon Signed Rank statistical analysis.
The mass spectrometer used was purchased via an NIH shared instrument grant, 1S10RR031537-01. This work is supported by funding from the National Institutes of Health: R01- AI104920 to J.G.S. and P.L.S., and U24 GM116788, S10 OD018142-01, and S10 RR25080-01 to the Southeastern Consortium for Microscopy of MacroMolecular Machines at Florida State University. We also acknowledge T32 GM008803 training grant support to N.M.G. M.E.W. was supported by the Helen Riaboff Whiteley Endowment and by the Public Health Service, National Research Service award T32 GM007270 from the National Institute of General Medical Sciences.
The authors report no relevant conflicts of interest.
1. Graham SV. The human papillomavirus replication cycle, and its links to cancer progression: a comprehensive review. Clin Sci (Lond). 2017;131(17):2201-21. PubMed PMID: 28798073. doi: 10.1042/CS20160786
2. Schiffman M, Doorbar J, Wentzensen N, de Sanjose S, Fakhry C, Monk BJ, Stanley MA, Franceschi S. Carcinogenic human papillomavirus infection. Nat Rev Dis Primers. 2016;2:16086. PubMed PMID: 27905473. doi: 10.1038/nrdp.2016.86
3. Orbegoso C, Murali K, Banerjee S. The current status of immunotherapy for cervical cancer. Rep Pract Oncol Radiother. 2018;23(6):580-8. PubMed PMID: 30534022. Pubmed Central PMCID: PMC6277269. doi: 10.1016/j.rpor.2018.05.001
4. World Health Organization. Human papillomavirus vaccines: WHO position paper, May 2017-Recommendations. Vaccine. 2017;35:5753-5.
5. Harper DM, DeMars LR. HPV vaccines - A review of the first decade. Gynecol Oncol. 2017;146(1):196-204. PubMed PMID: 28442134. doi: 10.1016/j.ygyno.2017.04.004
6. Schiller J, Lowy D. Explanations for the high potency of HPV prophylactic vaccines. Vaccine. 2018;36(32 Pt A):4768-73. PubMed PMID: 29325819. Pubmed Central PMCID: PMC6035892. doi: 10.1016/j.vaccine.2017.12.079
7. Hong S, Laimins LA. Manipulation of the innate immune response by human papillomaviruses. Virus Res. 2017;231:34-40. PubMed PMID: 27826042. Pubmed Central PMCID: PMC5325814. doi: 10.1016/j.virusres.2016.11.004
8. Holly MK, Diaz K, Smith JG. Defensins in Viral Infection and Pathogenesis. Annu Rev Virol. 2017. PubMed PMID: 28715972. doi: 10.1146/annurev-virology-101416-041734
9. Rehaume LM, Hancock RE. Neutrophil-derived defensins as modulators of innate immune function. Crit Rev Immunol. 2008;28(3):185-200. PubMed PMID: 19024344.
10. Yang D, Liu ZH, Tewary P, Chen Q, de la Rosa G, Oppenheim JJ. Defensin participation in innate and adaptive immunity. Curr Pharm Des. 2007;13(30):3131-9. PubMed PMID: 17979754.
11. Wilson SS, Wiens ME, Smith JG. Antiviral mechanisms of human defensins. J Mol Biol. 2013;425(24):4965-80. PubMed PMID: 24095897. Pubmed Central PMCID: PMC3842434. doi: 10.1016/j.jmb.2013.09.038
12. Lehrer RI, Lu W. alpha-Defensins in human innate immunity. Immunol Rev. 2012;245(1):84-112. PubMed PMID: 22168415. doi: 10.1111/j.1600-065X.2011.01082.x
13. Porter E, Yang H, Yavagal S, Preza GC, Murillo O, Lima H, Greene S, Mahoozi L, Klein-Patel M, Diamond G, Gulati S, Ganz T, Rice PA, Quayle AJ. Distinct defensin profiles in Neisseria gonorrhoeae and Chlamydia trachomatis urethritis reveal novel epithelial cell-neutrophil interactions. Infect Immun. 2005;73(8):4823-33. PubMed PMID: 16040996. Pubmed Central PMCID: PMC1201278. doi: 10.1128/IAI.73.8.4823-4833.2005
14. Quayle AJ, Porter EM, Nussbaum AA, Wang YM, Brabec C, Yip KP, Mok SC. Gene expression, immunolocalization, and secretion of human defensin-5 in human female reproductive tract. Am J Pathol. 1998;152(5):1247-58. PubMed PMID: 9588893. Pubmed Central PMCID: PMC1858596.
15. Spencer JD, Hains DS, Porter E, Bevins CL, DiRosario J, Becknell B, Wang H, Schwaderer AL. Human alpha defensin 5 expression in the human kidney and urinary tract. PLoS One. 2012;7(2):e31712. PubMed PMID: 22359618. Pubmed Central PMCID: PMC3281003. doi: 10.1371/journal.pone.0031712
16. Buck CB, Day PM, Thompson CD, Lubkowski J, Lu W, Lowy DR, Schiller JT. Human alpha-defensins block papillomavirus infection. Proc Natl Acad Sci USA. 2006;103(5):1516-21. PubMed PMID: 16432216. Pubmed Central PMCID: PMC1360544. doi: 10.1073/pnas.0508033103
17. Tenge VR, Gounder AP, Wiens ME, Lu W, Smith JG. Delineation of interfaces on human alpha-defensins critical for human adenovirus and human papillomavirus inhibition. PLoS Pathog. 2014;10(9):e1004360. PubMed PMID: 25188351. Pubmed Central PMCID: PMC4154873. doi: 10.1371/journal.ppat.1004360
18. Wiens ME, Smith JG. Alpha-defensin HD5 inhibits furin cleavage of human papillomavirus 16 L2 to block infection. J Virol. 2015;89(5):2866-74. PubMed PMID: 25540379. Pubmed Central PMCID: PMC4325740. doi: 10.1128/JVI.02901-14
19. Wiens ME, Smith JG. alpha-Defensin HD5 Inhibits Human Papillomavirus 16 Infection via Capsid Stabilization and Redirection to the Lysosome. MBio. 2017;8(1). PubMed PMID: 28119475. Pubmed Central PMCID: PMC5263252. doi: 10.1128/mBio.02304-16
20. Gounder AP, Wiens ME, Wilson SS, Lu W, Smith JG. Critical determinants of human alpha-defensin 5 activity against non-enveloped viruses. J Biol Chem. 2012;287(29):24554-62. PubMed PMID: 22637473. Pubmed Central PMCID: PMC3397880. doi: 10.1074/jbc.M112.354068
21. Nguyen EK, Nemerow GR, Smith JG. Direct evidence from single-cell analysis that human {alpha}-defensins block adenovirus uncoating to neutralize infection. J Virol. 2010;84(8):4041-9. PubMed PMID: 20130047. Pubmed Central PMCID: PMC2849482. doi: 10.1128/JVI.02471-09
22. Smith JG, Nemerow GR. Mechanism of adenovirus neutralization by Human alpha-defensins. Cell Host Microbe. 2008;3(1):11-9. PubMed PMID: 18191790. doi: 10.1016/j.chom.2007.12.001
23. Smith JG, Silvestry M, Lindert S, Lu W, Nemerow GR, Stewart PL. Insight into the mechanisms of adenovirus capsid disassembly from studies of defensin neutralization. PLoS Pathog. 2010;6(6):e1000959. PubMed PMID: 20585634. Pubmed Central PMCID: PMC2891831. doi: 10.1371/journal.ppat.1000959
24. Snijder J, Reddy VS, May ER, Roos WH, Nemerow GR, Wuite GJ. Integrin and defensin modulate the mechanical properties of adenovirus. J Virol. 2013;87(5):2756-66. PubMed PMID: 23269786. Pubmed Central PMCID: PMC3571403. doi: 10.1128/JVI.02516-12
25. Flatt JW, Kim R, Smith JG, Nemerow GR, Stewart PL. An intrinsically disordered region of the adenovirus capsid is implicated in neutralization by human alpha defensin 5. PLoS One. 2013;8(4):e61571. PubMed PMID: 23620768. Pubmed Central PMCID: PMC3631211. doi: 10.1371/journal.pone.0061571
26. Zins SR, Nelson CD, Maginnis MS, Banerjee R, O'Hara BA, Atwood WJ. The human alpha defensin HD5 neutralizes JC polyomavirus infection by reducing endoplasmic reticulum traffic and stabilizing the viral capsid. J Virol. 2014;88(2):948-60. PubMed PMID: 24198413. Pubmed Central PMCID: PMC3911681. doi: 10.1128/JVI.02766-13
27. Dugan AS, Maginnis MS, Jordan JA, Gasparovic ML, Manley K, Page R, Williams G, Porter E, O'Hara BA, Atwood WJ. Human alpha-defensins inhibit BK virus infection by aggregating virions and blocking binding to host cells. J Biol Chem. 2008;283(45):31125-32. PubMed PMID: 18782756. Pubmed Central PMCID: PMC2576552. doi: 10.1074/jbc.M805902200
28. Li M, Beard P, Estes PA, Lyon MK, Garcea RL. Intercapsomeric disulfide bonds in papillomavirus assembly and disassembly. J Virol. 1998;72(3):2160-7. PubMed PMID: 9499072. Pubmed Central PMCID: PMC109511.
29. Guan J, Bywaters SM, Brendle SA, Ashley RE, Makhov AM, Conway JF, Christensen ND, Hafenstein S. Cryoelectron Microscopy Maps of Human Papillomavirus 16 Reveal L2 Densities and Heparin Binding Site. Structure. 2017;25(2):253-63. PubMed PMID: 28065506. doi: 10.1016/j.str.2016.12.001
30. Cerqueira C, Pang YY, Day PM, Thompson CD, Buck CB, Lowy DR, Schiller JT. A Cell-Free Assembly System for Generating Infectious Human Papillomavirus 16 Capsids Implicates a Size Discrimination Mechanism for Preferential Viral Genome Packaging. J Virol. 2016;90(2):1096-107. PubMed PMID: 26559838. Pubmed Central PMCID: PMC4702664. doi: 10.1128/JVI.02497-15
31. Roden RB, Greenstone HL, Kirnbauer R, Booy FP, Jessie J, Lowy DR, Schiller JT. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. J Virol. 1996;70(9):5875-83. PubMed PMID: 8709207. Pubmed Central PMCID: PMC190605.
32. Bergant Marusic M, Ozbun MA, Campos SK, Myers MP, Banks L. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic. 2012;13(3):455-67. PubMed PMID: 22151726. Pubmed Central PMCID: PMC3276720. doi: 10.1111/j.1600-0854.2011.01320.x
33. Lipovsky A, Popa A, Pimienta G, Wyler M, Bhan A, Kuruvilla L, Guie MA, Poffenberger AC, Nelson CD, Atwood WJ, DiMaio D. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc Natl Acad Sci U S A. 2013;110(18):7452-7. PubMed PMID: 23569269. Pubmed Central PMCID: PMC3645514. doi: 10.1073/pnas.1302164110
34. Pim D, Broniarczyk J, Bergant M, Playford MP, Banks L. A Novel PDZ Domain Interaction Mediates the Binding between Human Papillomavirus 16 L2 and Sorting Nexin 27 and Modulates Virion Trafficking. J Virol. 2015;89(20):10145-55. PubMed PMID: 26202251. Pubmed Central PMCID: PMC4580170. doi: 10.1128/JVI.01499-15
35. Popa A, Zhang W, Harrison MS, Goodner K, Kazakov T, Goodwin EC, Lipovsky A, Burd CG, DiMaio D. Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog. 2015;11(2):e1004699. PubMed PMID: 25693203. Pubmed Central PMCID: PMC4334968. doi: 10.1371/journal.ppat.1004699
36. Zhang P, Monteiro da Silva G, Deatherage C, Burd C, DiMaio D. Cell-Penetrating Peptide Mediates Intracellular Membrane Passage of Human Papillomavirus L2 Protein to Trigger Retrograde Trafficking. Cell. 2018;174(6):1465-76 e13. PubMed PMID: 30122350. Pubmed Central PMCID: PMC6128760. doi: 10.1016/j.cell.2018.07.031
37. Zhang W, Kazakov T, Popa A, DiMaio D. Vesicular trafficking of incoming human papillomavirus 16 to the Golgi apparatus and endoplasmic reticulum requires gamma-secretase activity. MBio. 2014;5(5):e01777-14. PubMed PMID: 25227470. Pubmed Central PMCID: PMC4172078. doi: 10.1128/mBio.01777-14
38. Aydin I, Villalonga-Planells R, Greune L, Bronnimann MP, Calton CM, Becker M, Lai KY, Campos SK, Schmidt MA, Schelhaas M. A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLoS Pathog. 2017;13(5):e1006308. PubMed PMID: 28464022. Pubmed Central PMCID: PMC5412989. doi: 10.1371/journal.ppat.1006308
39. Calton CM, Bronnimann MP, Manson AR, Li S, Chapman JA, Suarez-Berumen M, Williamson TR, Molugu SK, Bernal RA, Campos SK. Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLoS Pathog. 2017;13(5):e1006200. PubMed PMID: 28463988. Pubmed Central PMCID: PMC5412990. doi: 10.1371/journal.ppat.1006200
40. Buck CB, Cheng N, Thompson CD, Lowy DR, Steven AC, Schiller JT, Trus BL. Arrangement of L2 within the papillomavirus capsid. J Virol. 2008;82(11):5190-7. PubMed PMID: 18367526. Pubmed Central PMCID: PMC2395198. doi: 10.1128/JVI.02726-07
41. Bywaters SM, Brendle SA, Tossi KP, Biryukov J, Meyers C, Christensen ND. Antibody Competition Reveals Surface Location of HPV L2 Minor Capsid Protein Residues 17-36. Viruses. 2017;9(11). PubMed PMID: 29125554. Pubmed Central PMCID: PMC5707543. doi: 10.3390/v9110336
42. Kondo K, Ishii Y, Ochi H, Matsumoto T, Yoshikawa H, Kanda T. Neutralization of HPV16, 18, 31, and 58 pseudovirions with antisera induced by immunizing rabbits with synthetic peptides representing segments of the HPV16 minor capsid protein L2 surface region. Virology. 2007;358(2):266-72. PubMed PMID: 17010405. doi: 10.1016/j.virol.2006.08.037
43. Pastrana DV, Gambhira R, Buck CB, Pang YY, Thompson CD, Culp TD, Christensen ND, Lowy DR, Schiller JT, Roden RB. Cross-neutralization of cutaneous and mucosal Papillomavirus types with anti-sera to the amino terminus of L2. Virology. 2005;337(2):365-72. PubMed PMID: 15885736. doi: 10.1016/j.virol.2005.04.011
44. Buck CB, Pastrana DV, Lowy DR, Schiller JT. Efficient intracellular assembly of papillomaviral vectors. J Virol. 2004;78(2):751-7. PubMed PMID: 14694107. Pubmed Central PMCID: PMC368835.
45. Buck CB, Thompson CD. Production of papillomavirus-based gene transfer vectors. Curr Protoc Cell Biol. 2007;Chapter 26:Unit 26 1. PubMed PMID: 18228512. doi: 10.1002/0471143030.cb2601s37
46. Buck CB, Thompson CD, Pang YY, Lowy DR, Schiller JT. Maturation of papillomavirus capsids. J Virol. 2005;79(5):2839-46. PubMed PMID: 15709003. Pubmed Central PMCID: PMC548454. doi: 10.1128/JVI.79.5.2839-2846.2005
47. Cardone G, Moyer AL, Cheng N, Thompson CD, Dvoretzky I, Lowy DR, Schiller JT, Steven AC, Buck CB, Trus BL. Maturation of the human papillomavirus 16 capsid. MBio. 2014;5(4):e01104-14. PubMed PMID: 25096873. Pubmed Central PMCID: PMC4128349. doi: 10.1128/mBio.01104-14
48. Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157(1):38-46. PubMed PMID: 16859925. doi: 10.1016/j.jsb.2006.05.009
49. Zhang K. Gctf: Real-time CTF determination and correction. J Struct Biol. 2016;193(1):1-12. PubMed PMID: 26592709. Pubmed Central PMCID: PMC4711343. doi: 10.1016/j.jsb.2015.11.003
50. Shevchenko A, Tomas H, Havliš J, Olsen JV, Mann M. In-Gel Digestion for Mass Spectrometric Characterization of Proteins and Proteomes. Nat Protoc. 2007;1(6):2856–60.
51. Kadiyala CS, Zheng L, Du Y, Yohannes E, Kao HY, Miyagi M, Kern TS. Acetylation of retinal histones in diabetes increases inflammatory proteins: effects of minocycline and manipulation of histone acetyltransferase (HAT) and histone deacetylase (HDAC). J Biol Chem. 2012;287(31):25869-80. PubMed PMID: 22648458. Pubmed Central PMCID: PMC3406672. doi: 10.1074/jbc.M112.375204
52. Vukoti K, Yu X, Sheng Q, Saha S, Feng Z, Hsu AL, Miyagi M. Monitoring newly synthesized proteins over the adult life span of Caenorhabditis elegans. J Proteome Res. 2015;14(3):1483-94. PubMed PMID: 25686393. Pubmed Central PMCID: PMC4440835. doi: 10.1021/acs.jproteome.5b00021
53. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605-12. PubMed PMID: 15264254. doi: 10.1002/jcc.20084
54. Kaplan W, Littlejohn TG. Swiss-PDB Viewer (Deep View). Brief Bioinform. 2001;2(2):195-7. PubMed PMID: 11465736. doi: 10.1093/bib/2.2.195
55. Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50(3):437-50. PubMed PMID: 12557186. doi: 10.1002/prot.10286
56. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781-802. PubMed PMID: 16222654. Pubmed Central PMCID: PMC2486339. doi: 10.1002/jcc.20289
57. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33-8, 27-8. PubMed PMID: 8744570.
58. Merkley ED, Rysavy S, Kahraman A, Hafen RP, Daggett V, Adkins JN. Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci. 2014;23(6):747-59. PubMed PMID: 24639379. Pubmed Central PMCID: PMC4093951. doi: 10.1002/pro.2458
59. Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180(3):519-30. PubMed PMID: 23000701. Pubmed Central PMCID: PMC3690530. doi: 10.1016/j.jsb.2012.09.006
60. Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J Virol. 2012;86(18):9875-87. PubMed PMID: 22761365. Pubmed Central PMCID: PMC3446629. doi: 10.1128/JVI.00980-12
61. Day PM, Thompson CD, Schowalter RM, Lowy DR, Schiller JT. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J Virol. 2013;87(7):3862-70. PubMed PMID: 23345514. Pubmed Central PMCID: PMC3624235. doi: 10.1128/JVI.03222-12
62. DiGiuseppe S, Bienkowska-Haba M, Guion LGM, Keiffer TR, Sapp M. Human Papillomavirus Major Capsid Protein L1 Remains Associated with the Incoming Viral Genome throughout the Entry Process. J Virol. 2017;91(16). PubMed PMID: 28566382. Pubmed Central PMCID: PMC5533910. doi: 10.1128/JVI.00537-17
63. de Leeuw E, Rajabi M, Zou G, Pazgier M, Lu W. Selective arginines are important for the antibacterial activity and host cell interaction of human alpha-defensin 5. FEBS Lett. 2009;583(15):2507-12. PubMed PMID: 19589339. doi: 10.1016/j.febslet.2009.06.051
64. Rajabi M, Ericksen B, Wu X, de Leeuw E, Zhao L, Pazgier M, Lu W. Functional determinants of human enteric alpha-defensin HD5: crucial role for hydrophobicity at dimer interface. J Biol Chem. 2012;287(26):21615-27. PubMed PMID: 22573326. Pubmed Central PMCID: PMC3381126. doi: 10.1074/jbc.M112.367995
65. Wang C, Shen M, Gohain N, Tolbert WD, Chen F, Zhang N, Yang K, Wang A, Su Y, Cheng T, Zhao J, Pazgier M, Wang J. Design of a potent antibiotic peptide based on the active region of human defensin 5. J Med Chem. 2015;58(7):3083-93. PubMed PMID: 25782105. doi: 10.1021/jm501824a
66. Xu D, Liao C, Zhang B, Tolbert WD, He W, Dai Z, Zhang W, Yuan W, Pazgier M, Liu J, Yu J, Sansonetti PJ, Bevins CL, Shao Y, Lu W. Human Enteric alpha-Defensin 5 Promotes Shigella Infection by Enhancing Bacterial Adhesion and Invasion. Immunity. 2018;48(6):1233-44 e6. PubMed PMID: 29858013. Pubmed Central PMCID: PMC6051418. doi: 10.1016/j.immuni.2018.04.014
67. Richards RM, Lowy DR, Schiller JT, Day PM. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc Natl Acad Sci U S A. 2006;103(5):1522-7. PubMed PMID: 16432208. Pubmed Central PMCID: PMC1360554. doi: 10.1073/pnas.0508815103
68. Bienkowska-Haba M, Patel HD, Sapp M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009;5(7):e1000524. PubMed PMID: 19629175. Pubmed Central PMCID: PMC2709439. doi: 10.1371/journal.ppat.1000524
69. Bronnimann MP, Calton CM, Chiquette SF, Li S, Lu M, Chapman JA, Bratton KN, Schlegel AM, Campos SK. Furin Cleavage of L2 during Papillomavirus Infection: Minimal Dependence on Cyclophilins. J Virol. 2016;90(14):6224-34. PubMed PMID: 27122588. Pubmed Central PMCID: PMC4936150. doi: 10.1128/JVI.00038-16
70. Day PM, Gambhira R, Roden RB, Lowy DR, Schiller JT. Mechanisms of human papillomavirus type 16 neutralization by l2 cross-neutralizing and l1 type-specific antibodies. J Virol. 2008;82(9):4638-46. PubMed PMID: 18305047. Pubmed Central PMCID: PMC2293042. doi: 10.1128/JVI.00143-08
71. Rubio I, Seitz H, Canali E, Sehr P, Bolchi A, Tommasino M, Ottonello S, Muller M. The N-terminal region of the human papillomavirus L2 protein contains overlapping binding sites for neutralizing, cross-neutralizing and non-neutralizing antibodies. Virology. 2011;409(2):348-59. PubMed PMID: 21074234. doi: 10.1016/j.virol.2010.10.017
72. Yang R, Day PM, Yutzy WHt, Lin KY, Hung CF, Roden RB. Cell surface-binding motifs of L2 that facilitate papillomavirus infection. J Virol. 2003;77(6):3531-41. PubMed PMID: 12610128. Pubmed Central PMCID: PMC149523.
73. Heino P, Skyldberg B, Lehtinen M, Rantala I, Hagmar B, Kreider JW, Kirnbauer R, Dillner J. Human papillomavirus type 16 capsids expose multiple type-restricted and type-common antigenic epitopes. J Gen Virol. 1995;76 ( Pt 5):1141-53. PubMed PMID: 7537325. doi: 10.1099/0022-1317-76-5-1141
74. Szyk A, Wu Z, Tucker K, Yang D, Lu W, Lubkowski J. Crystal structures of human alpha-defensins HNP4, HD5, and HD6. Protein Sci. 2006;15(12):2749-60. PubMed PMID: 17088326. Pubmed Central PMCID: PMC2242434. doi: 10.1110/ps.062336606
75. Bullock JMA, Sen N, Thalassinos K, Topf M. Modeling Protein Complexes Using Restraints from Crosslinking Mass Spectrometry. Structure. 2018;26(7):1015-24 e2. PubMed PMID: 29804821. Pubmed Central PMCID: PMC6039719. doi: 10.1016/j.str.2018.04.016
76. Campos SK. Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2. Viruses. 2017;9(12). PubMed PMID: 29207511. Pubmed Central PMCID: PMC5744145. doi: 10.3390/v9120370
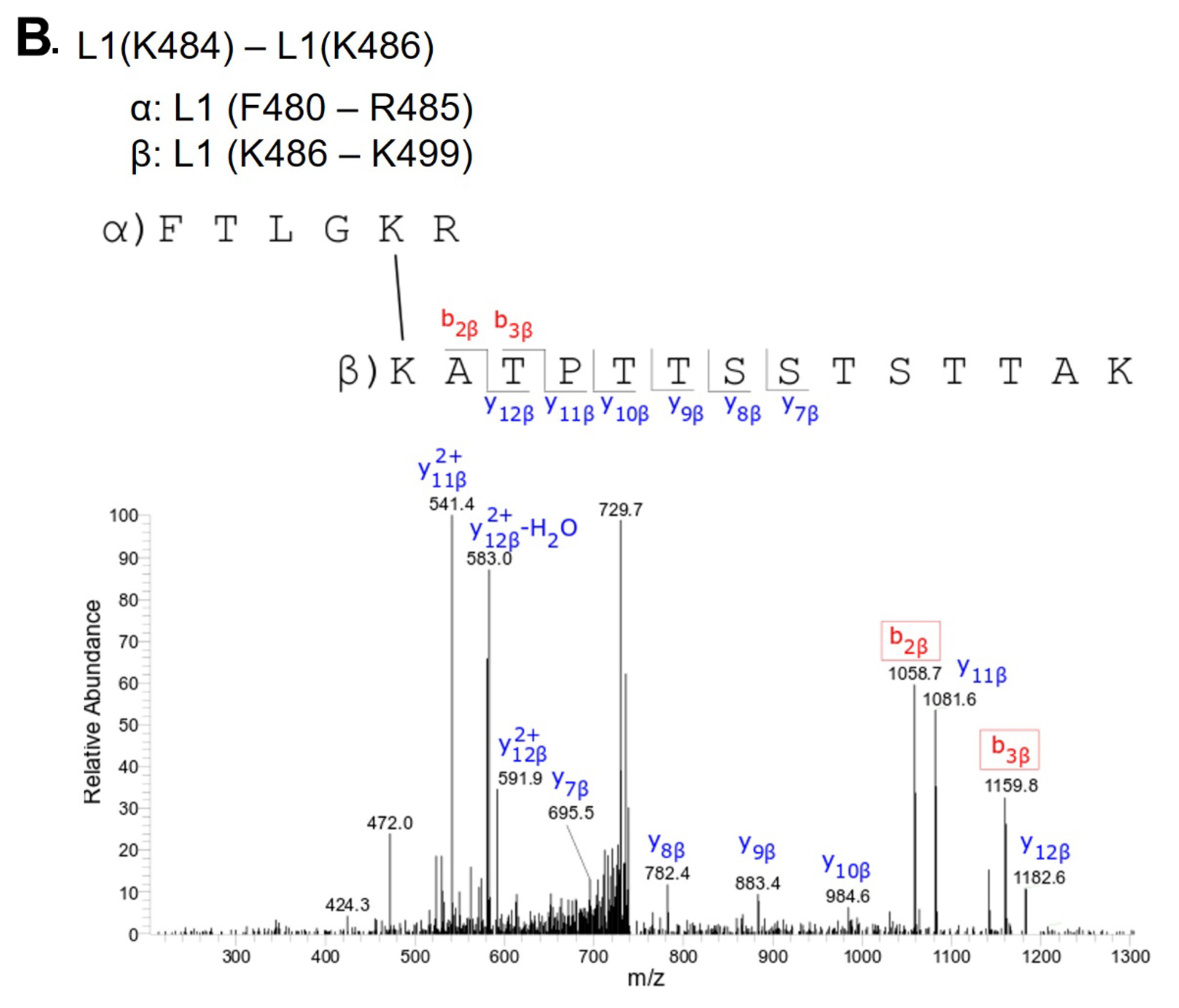
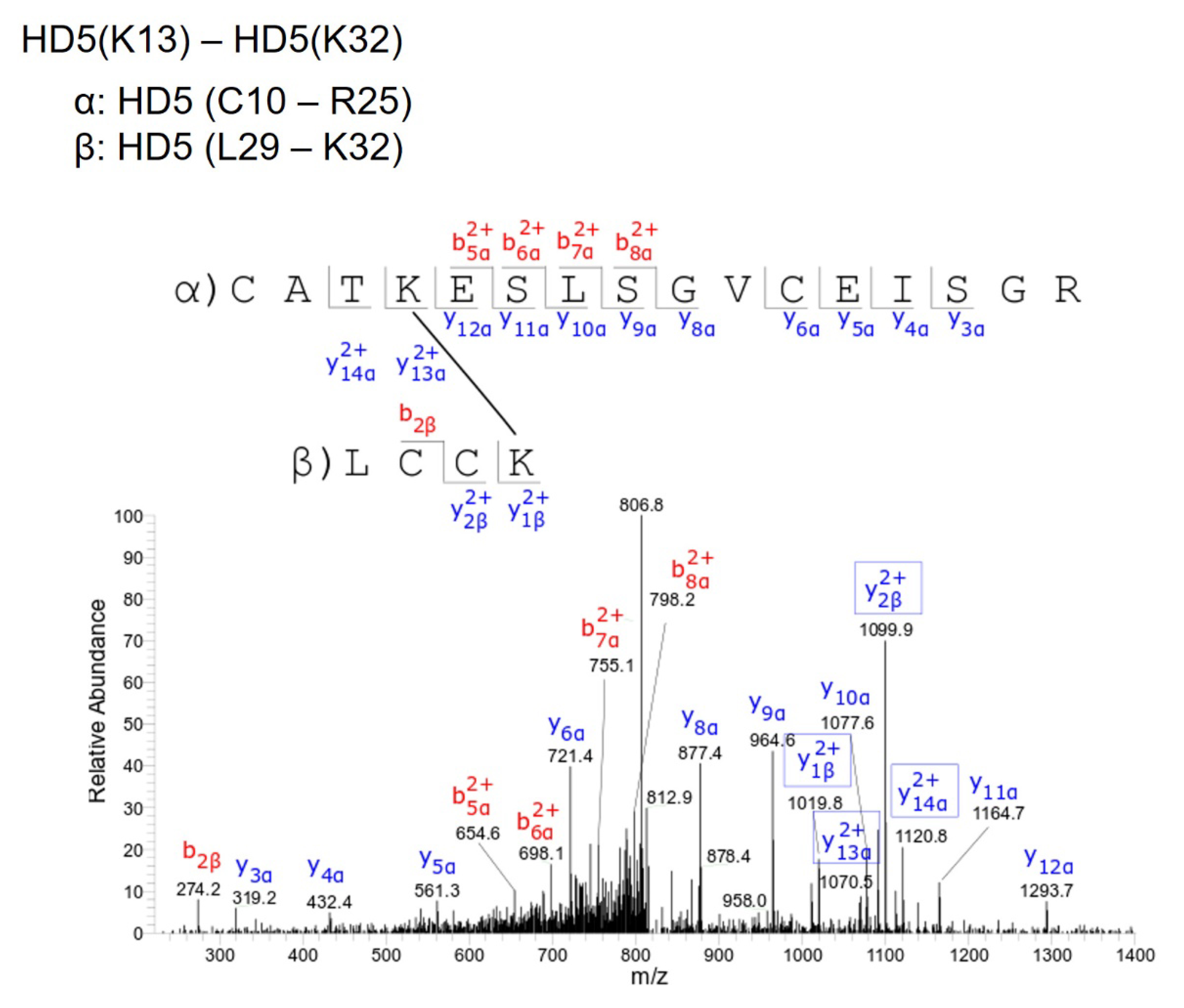

Supplementary Table 1. Mass spectrometry detected BS3 intra-protein crosslinks within L1
|
BS3 crosslinked residues |
Band position on gel |
|
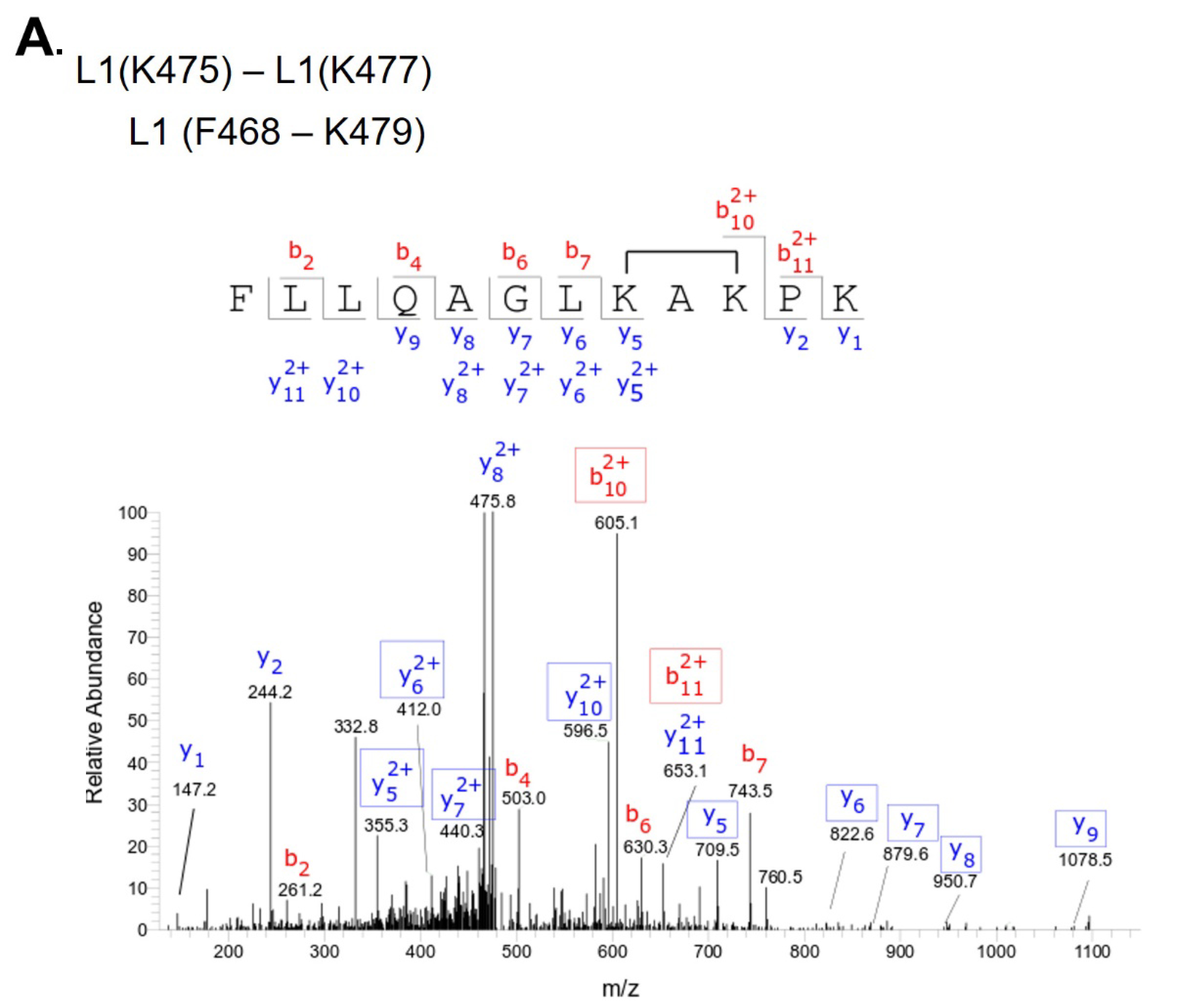
L1(K475) - L1(K477) |
Above L1 band, and L1/L2 dimer region |
|
L1(K484) - L1(K486) |
Above L1 band, and L1/L2 dimer region |
Supplementary Table 2. Mass spectrometry detected BS3 intra-protein crosslinks within HD5-R13K/R32K
|
BS3 crosslinked residues |
Band position on gel |
|
HD5(K13) - HD5(K32) |
Above L1 band, and above L2 band |